Unusual Manifestation of a Rare Disorder: Type XIV Osteogenesis Imperfecta Presenting as Fetal Hydrops
Roopadarshini B1, Sreeja P1, Ashwin Dalal2, Prajnya Ranganath1 1 Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad, India 2 Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India Correspondence to: Dr Prajnya RanganathEmail:prajnyaranganath@gmail.com
1 Abstract
Osteogenesis imperfecta (OI) is a group of inherited disorders characterised by decreased bone density and increased
susceptibility to fractures. In addition to the classic COL1A1/ COL1A2- associated autosomal dominant (AD) OI, close
to 20 more types have been identified in recent years. Type XIV OI is a very rare autosomal recessive form of OI caused
by biallelic variants in the TMEM38B gene. We report here a consanguineous family with recurrent fetal hydrops,
where evaluation of the second affected fetus revealed the diagnosis of TMEM38B gene-related Type XIV
OI.
Osteogenesis imperfecta (OI) is a clinically and genetically heterogeneous group of disorders of bone mineralization. Based
on the clinical manifestations, Sillence et al. (1979) classified OI into four main types namely the classic non-deforming
type with blue sclerae (type I OI), the perinatally lethal form (type II OI), the progressively deforming type (type III OI)
and the common variable type with normal sclerae (type IV OI). OI is classically associated with heterozygous variants in
the COL1A1 or COL1A2 genes which code for the α1 and α2 chains of collagen type I respectively. COL1A1/ COL1A2-
related OI has an autosomal dominant pattern of inheritance. Over the past one to two decades, following
the availability of high throughput molecular analysis techniques especially next-generation sequencing,
several more types of OIs due to variants in other genes involved in collagen I processing and/ or osteoblast
function have been identified. Majority of these have an autosomal recessive (AR) pattern of inheritance. One
such AR OI is type XIV OI (OMIM # 615066) which was first described in 2012 (Shaheen et al., 2012). It
is caused by biallelic variants in the TMEM38B gene (OMIM *611236) which codes for an endoplasmic
reticulum membrane channel called the trimeric intracellular cation channel type B (TRIC-B) (Ramzan
et al., 2021). It is a rare form of OI with variable degree of severity of fractures and osteopenia. Limited
case reports on type XIV are available and presentation with intrauterine hydrops has not been reported
previously.
3 Clinical Report
This 24-year-old second gravida, married consanguineously to her first cousin, presented at 20 weeks gestation with
antenatal scan findings of nuchal edema, generalized subcutaneous edema, shortening of all the long bones and bilateral
bent femora in the fetus. Her blood group was B positive. The first pregnancy of the couple had been terminated due to
similar antenatal scan findings; however, fetal autopsy and/ or genetic evaluation had not been done for the first
pregnancy. The couple opted for termination of this second affected pregnancy. Prior to termination, amniocentesis was
done, and the sample was sent for karyotyping and DNA extraction and storage. Following termination, the fetus was
submitted for autopsy evaluation.
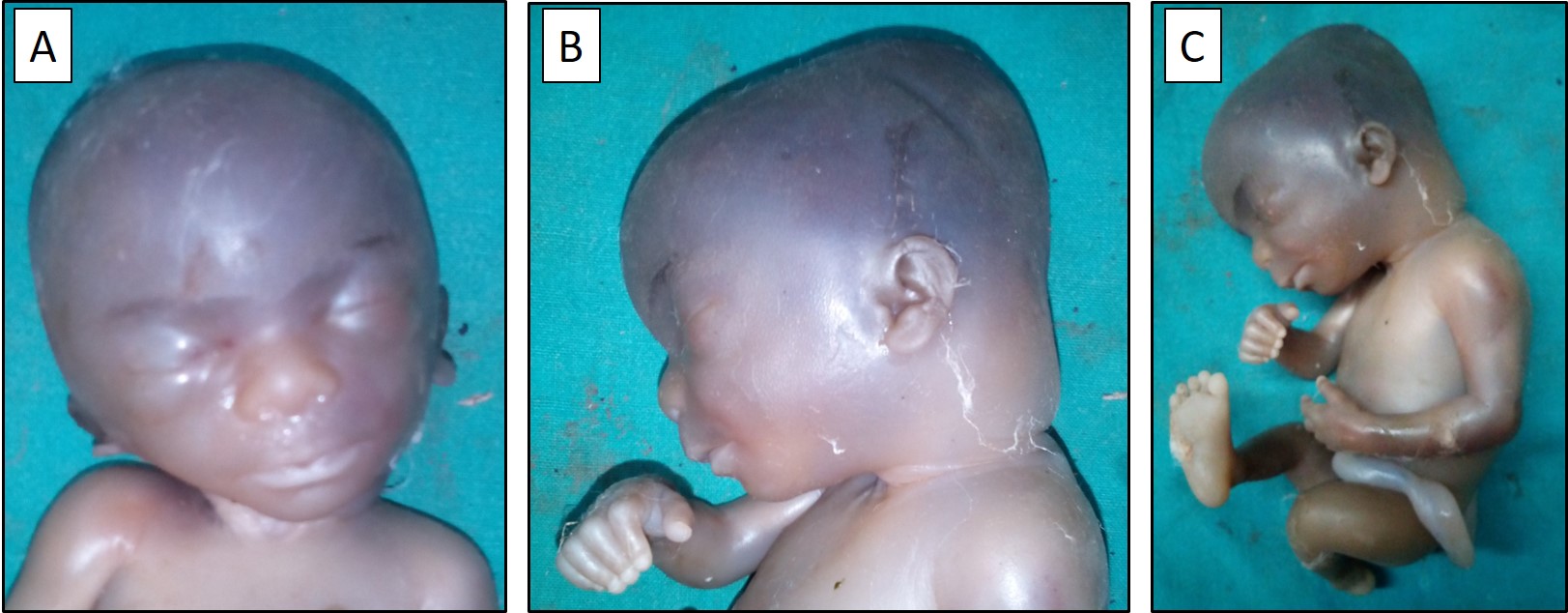
Figure 1A & 1B. Frontal and lateral views of the fetal head and face showing the scalp edema, nuchal
edema, and craniofacial dysmorphism in the form of a globular head shape, depressed nasal bridge, broad nose,
low-set ears and microretrognathia. 1C. Lateral view of the fetal body showing the generalized subcutaneous
edema.
On autopsy, the fetus was found to have a body weight of 350 g (˜70th centile), crown-heel length of 22 cm (8th
centile), crown-rump length of 15 cm (7th centile), foot length of 3 cm (2nd centile), head circumference of 17.5 cm (56th
centile) and chest circumference of 14.5 cm (60th centile). Craniofacial dysmorphism was noted in the form of a globular
head shape, depressed nasal bridge, broad nose, low-set ears and microretrognathia (Figure 1A & 1B). There was scalp
edema, nuchal edema, and generalised subcutaneous edema (Figure 1B & 1C). On external examination, the chest and
back were normal, and the abdomen appeared to be distended. The thighs appeared short and bent. Other
limb segments appeared to be proportionate and symmetric. Bilateral upper limb measurements were as
follows: proximal segment 4 cm, middle segment 3.5 cm, and distal segment (hand) 2 cm. Bilateral lower limb
measurements were as follows: proximal segment 4 cm, middle segment 4 cm, and distal segment (foot)
3 cm. Normal male external genitalia and normal anal opening were noted. Internal dissection revealed
bilateral pleural effusion and ascites. The intrathoracic and intraabdominal organs including the heart, lungs,
great vessels, stomach, small and large intestines, liver, and spleen appeared to be grossly normal. The
kidneys and the urinary tract also appeared to be normal. The placenta was normal, and the cord had three
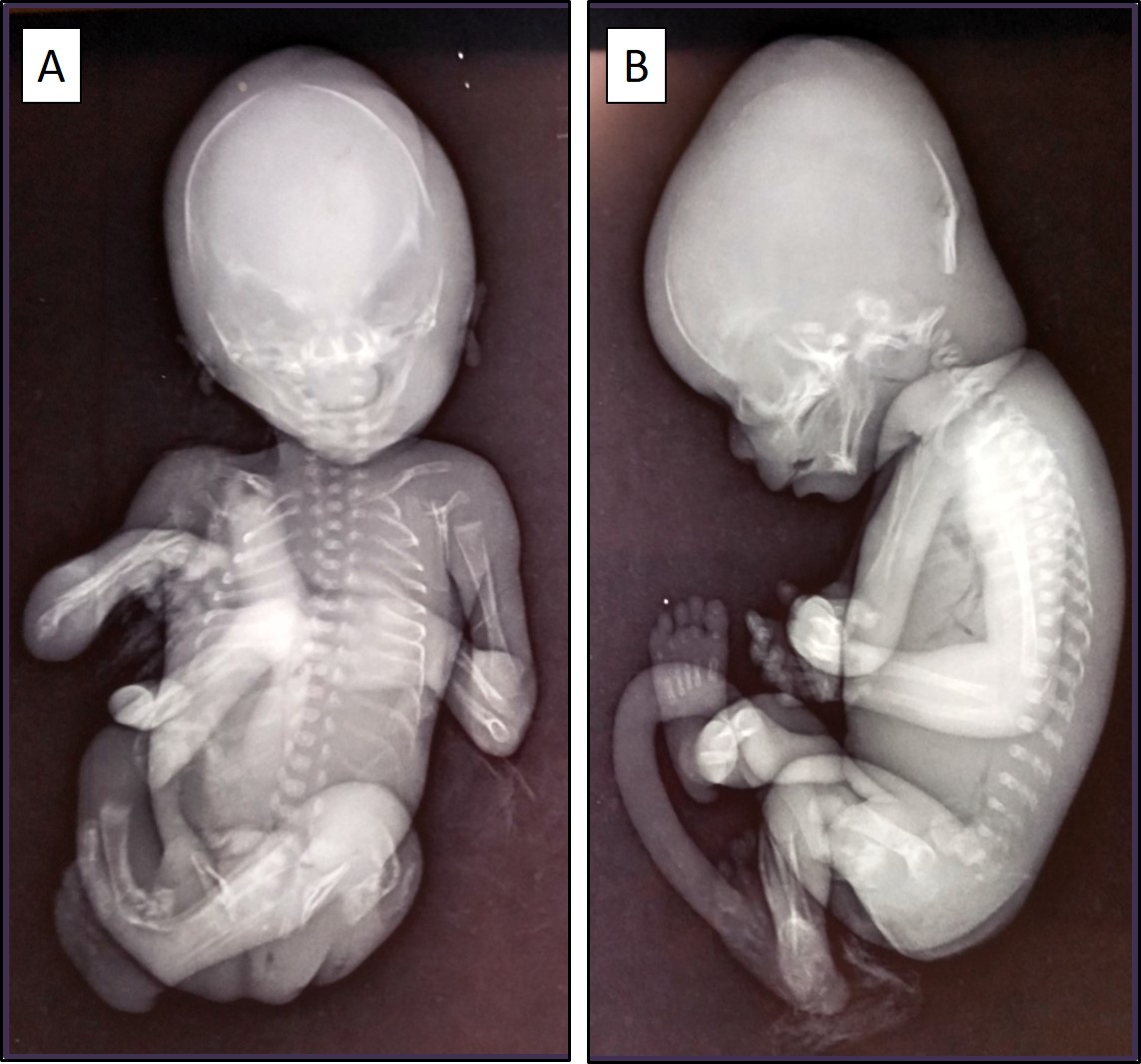
vessels. Fetal skeletal radiographs showed bent and shortened femora with acute angulation in the femoral
shaft bilaterally. The ribs appeared to be thin and wavy, and poor mineralization of the long bones and
cranium was noted (Figure 2). Histopathological evaluation of the placenta and the intrathoracic and
intra-abdominal organs did not reveal any significant abnormality. Based on the clinical and radiographic findings, the
possibility of a skeletal dysplasia with poor bone mineralization especially osteogenesis imperfecta was
considered.
Figure 2A & 2B. Fetal skeletal radiographs (anteroposterior and lateral views) showing bent and
shortened femora, thin and wavy ribs, and poor mineralization of the long bones and cranium.
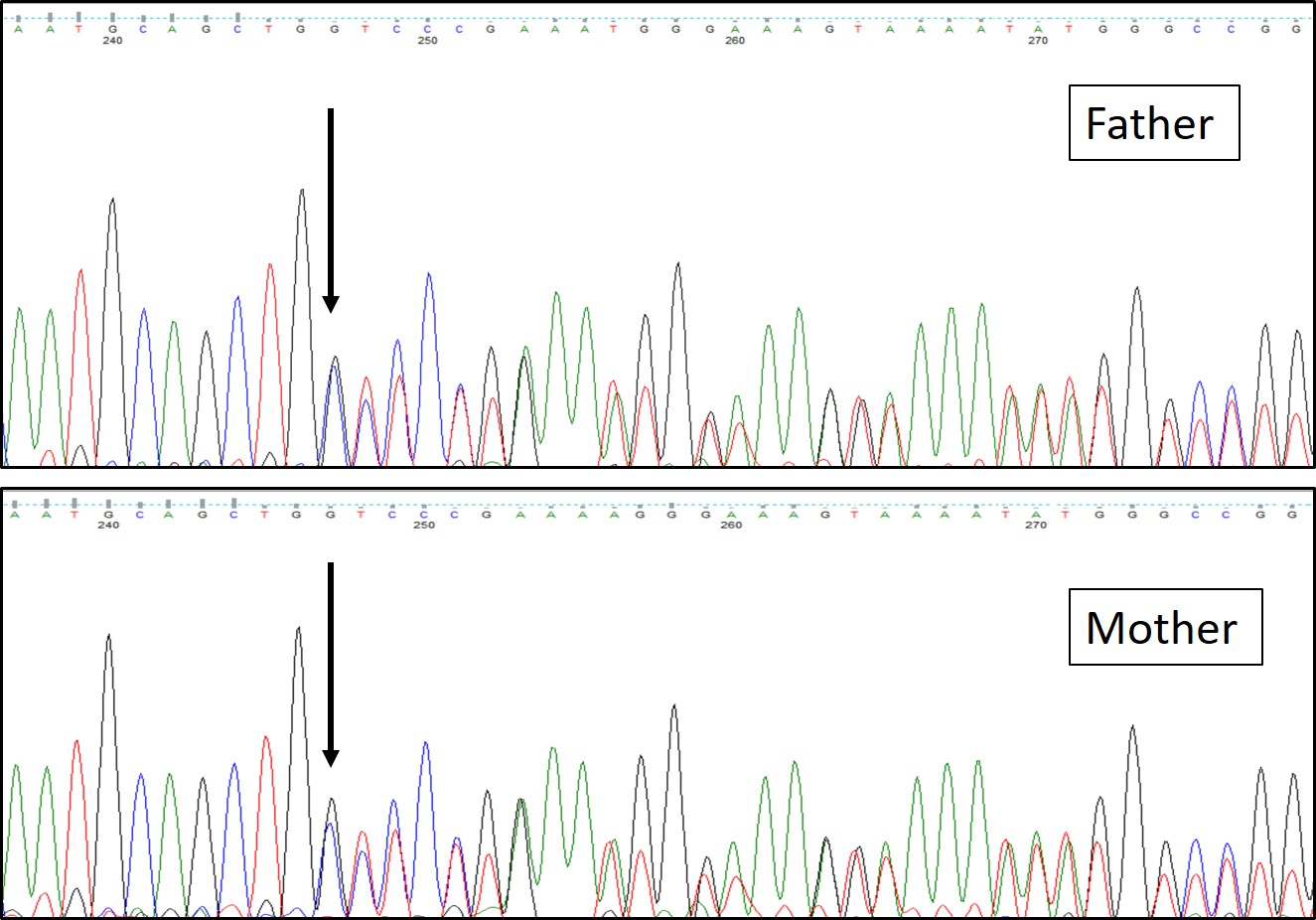
Figure 3. Targeted Sanger sequencing of both parents showing heterozygous carrier status for the
variant c.115_116insC (p.Ala40SerfsTer43) in the TMEM38B gene (ENST00000374692).
Karyotype of the amniotic fluid was reported to be normal. In view of the likely diagnosis of osteogenesis imperfecta
based on the fetal autopsy findings, whole-exome sequencing (WES) was done in the stored fetal DNA, which revealed a
homozygous novel frameshift variant c.115_116insC (p.Ala40SerfsTer43) in the TMEM38B gene (ENST00000374692).
The variant is absent in the population databases gnomAD ( https://gnomad.broadinstitute.org/) and 1000
Genomes ( https://www.internationalgenome.org/1000-genomes-browsers/). It is classified as a ‘likely
pathogenic’ variant (based on the criteria PVS1 + PM2) as per the variant classification guidelines of the American
College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015). Through
targeted Sanger sequencing, both parents were confirmed to be heterozygous carriers of this variant (Figure3).
Based on the autopsy and molecular genetic findings, the diagnosis for the fetus was concluded to be TMEM38B
gene-related type XIV osteogenesis imperfecta with non-immune fetal hydrops.
4 Discussion
Type XIV OI was first described by Shaheen and co-workers in 2012 (Shaheen et al., 2012). They described 12 affected
individuals from three families in Saudi Arabia with clinical symptoms of OI. By autozygosity mapping and linkage
analysis they found a novel recessive OI locus which mapped to chromosome 9q31.1-31.3. They identified a novel
truncating deletion of exon 4 in the TMEM38B gene within that locus. The same exonic deletion was reported by
Volodarsky and group in affected members of three unrelated Israeli Bedouin consanguineous families (Volodarsky et al.,
2013). At present, 17 mutations in the TMEM38B gene are listed in the Human Gene Mutation Database (HGMD;
https://www.hgmd.cf.ac.uk/).
TMEM38B codes for trimeric intracellular cation channel type B which is present in the endoplasmic reticulum. This
cation channel regulates intracellular calcium influx. The role of fine-tuned intracellular calcium levels in the proliferation,
differentiation, and cellular function of numerous cell types including osteoblasts has been clearly established (Berridge
et.al, 2000; Zayzafoon, 2006).
The clinical manifestations and molecular basis of type XIV OI were studied in depth by Webb and coworkers (Webb
et al., 2017). They studied eight patients with type XIV OI and in addition to the usual manifestations of OI, they
described previously unreported features like periosteal cloaking, coxa vara and extra-skeletal manifestations like muscular
hypotonia and cardiac abnormalities. They analysed bone biopsy samples of the patients and demonstrated decreased
trabecular bone volume as well as reduction of osteoblast and osteoclast numbers with more than 80% reduction in bone
resorption. They concluded that in addition to an intrinsic osteoclast defect leading to low bone turnover, there are
intracellular calcium flux abnormalities in type XIV OI which possibly cause the muscular and cardiovascular
features seen in this condition. Thus, type XIV OI has a distinctive pathogenesis from most other forms
of OI which are associated with defects in the formation, folding, or posttranslational modifications of
collagen.
Published literature related to type XIV OI is limited and most reported cases have had postnatal onset of
manifestations. Recently, Kodama and group published a case report of a newborn with multiple fractures of intrauterine
onset caused by a novel splice variant in the TMEM38B gene (Kodama et al., 2023). However, presentation of type XIV
OI as fetal hydrops has not been previously reported. Though the exact pathogenetic mechanism for the fetal
hydrops in our case remains to be established, it is possible that the calcium influx defects resulting from
biallelic variants in the TMEM38B gene may have led to congestive cardiac failure and third-space fluid
accumulation.
5 Conclusion
Type XIV OI is a rare autosomal recessive type of osteogenesis imperfecta, which has a molecular mechanism different
from that of the conventional collagen-related types of OI. Ours is the first case of type XIV OI to be reported with the
phenotype of hydrops fetalis, to the best of our knowledge. Further studies are expected to expand the phenotypic
spectrum of this rare form of OI.
Conflict of Interests: None
Acknowledgments: The authors thank the family for their participation in the study. Molecular genetic testing in
this study was performed with funding support obtained from the Science and Engineering Research Board (SERB),
Department of Science and Technology (DST), Government of India through the project EMR/2016/003887 granted to
the PI Dr Prajnya Ranganath.
References
1. Berridge MJ, et al. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol. 2000;
1(1): 11-21.
2. Kodama Y, et al. Novel splice site variant of TMEM38B in osteogenesis imperfecta type XIV. Hum Genome
Var. 2023;10(1): 25.
3. Ramzan K, et al. Detection of a recurrent TMEM38B gene deletion associated with recessive osteogenesis
imperfecta. Discoveries (Craiova). 2021; 9(1): e124.
4. Richards S, et al. & ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the
interpretation of sequence variants: a joint consensus recommendation of the American College of Medical
Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17(5): 405-424.
5. Shaheen R, et al. A study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus
defined by TMEM38B mutation. J Med Genet. 2012; 49(10): 630-635.
6. Sillence D, et al. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979; 16(2): 101-116.
7. Volodarsky M, et al. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis
imperfecta. Hum Mutat. 2013; 34(4): 582-586.
8. Webb EA, et al. Phenotypic spectrum in osteogenesis imperfecta due to mutations in TMEM38B:
Unraveling a complex cellular defect. J Clin Endocrinol Metab. 2017;102(6): 2019-2028.
9. Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem.
2006; 97(1): 56-70.