Ghosal Hematodiaphyseal Dysplasia: An Unusual but Easy-to-Diagnose Genetic Cause of Anemia
Amrita Bhattacherjee1, Ashwin B Dalal1, Prajnya Ranganath1,2,* 1Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India 2Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad, India Correspondence to: Dr Prajnya RanganathEmail:prajnyaranganath@gmail.com
1 Introduction
Ghosal hematodiaphyseal dysplasia (GHDD) also known as Ghosal syndrome (OMIM# 231095) is a rare autosomal
recessive disorder associated with skeletal changes in the form of increased bone density and predominant diaphyseal
involvement, and hypoplastic anemia (Ghosal et al., 1988). It is caused by biallelic variants in the TBXAS1
(OMIM*274180) gene. As the hematological abnormalities respond to corticosteroid therapy, early and accurate diagnosis
helps in effective control of the disease course. We report here a 12 years-old male patient from a consanguineous family
referred for evaluation of chronic anemia, whose clinical and radiographic findings were suggestive of the diagnosis of
GHDD and molecular genetic testing revealed a homozygous nonsynonymous known variant in the TBXAS1
gene.
2 Patient details
A 12-years-old boy, the third offspring of third-degree consanguineous parents, was referred for evaluation of chronic
anemia detected at around 6 years of age. He had received six transfusions of packed red blood cells (PRBC) between 6 to
12 years of age. There was no history of abdominal distension, jaundice, gall stones, bleeding manifestations, or recurrent
infections and fever. No specific symptoms were noted in early childhood. The developmental milestones were
age-appropriate but the scholastic performance was below average. The two elder female siblings aged 18 years and 15
years, and both parents were normal. There was no history of similar illness or any other known genetic disease in other
family members. On examination, his height and weight were 146 cm and 36 kgs respectively (both corresponding to
around 30th centile for age), and head circumference was 55 cm. He had severe pallor, but no icterus, edema, or
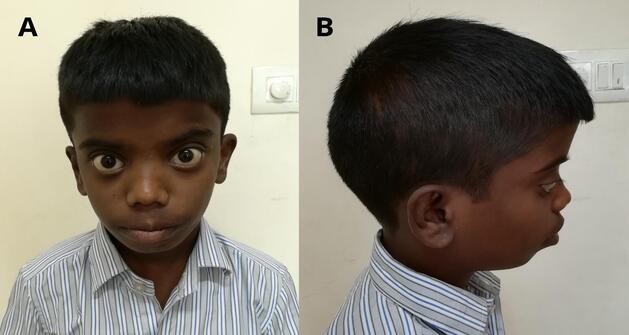
lymphadenopathy. Craniofacial dysmorphic features noted in the child included dolichocephaly, frontal
prominence, telecanthus, bilateral proptosis, depressed nasal bridge with bulbous tip of nose, thick lips, and
retrognathia (Figure 1). There was no hepatosplenomegaly and rest of the systemic examination was also
normal.
Figure 1: Photographs of the patient showing the craniofacial dysmorphic features. A. Frontal view showing
telecanthus, bilateral proptosis, depressed nasal bridge, bulbous nose and thick lips. B. Lateral view showing
dolichocephaly, depressed nasal bridge and retrognathia.
Hemogram revealed normocytic normochromic anemia (hemoglobin: 6.9 g/dL; red blood cell count: 2.62 × 106∕μL)
with normal total leucocyte count (5600/cu mm) and normal platelet count (3 × 105/cu mm). Bone marrow studies
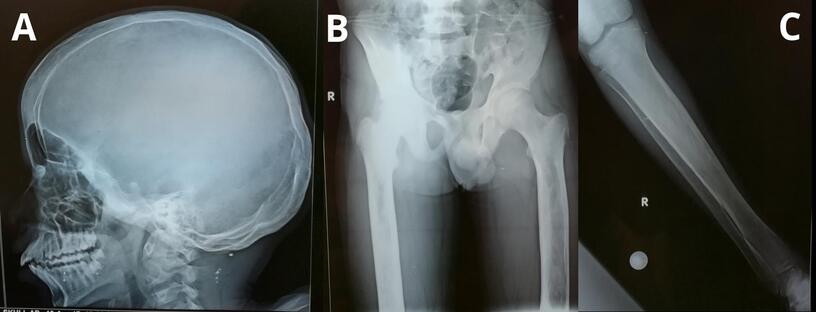
(aspiration and biopsy) showed evidence of marrow hypocellularity. Skeletal radiographs showed mild increase in bone
density, widening of the diploic spaces of the skull, sclerosis of the base of the skull, and mild diaphyseal widening and
cortical hyperostosis of the femoral and tibial bones bilaterally (Figure 2). Ophthalmological evaluation revealed bilateral
pseudo-proptosis with lagophthalmos. The salient findings of hypoplastic anemia and skeletal dysplastic
changes suggested the diagnosis of Ghosal hematodiaphyseal dysplasia. Though the clinical findings also
suggested the possibility of pycnodysostosis, the skeletal radiographic findings were not in favour of the
diagnosis.
Figure 2: Skeletal radiographs of the patient. A. Lateral view of the skull showing the widening of the diploic
spaces and sclerosis of the base of the skull. B. & C. Anteroposterior view of the pelvis, both femurs and the
right tibia & fibula showing the increased bone density, diaphyseal widening and cortical hyperostosis.
Next generation sequencing-based Exome sequencing was performed to look for variants in the TBXAS1 gene
(which has 17 exons of which 13 are coding exons) and to rule out variants in other genes associated with
diaphyseal dysplasia and sclerosing bone disorders (including Camurati-Engelmann disease-associated TGFB1
gene) and genes associated with bone marrow hypocellularity. A homozygous non-synonymous variant
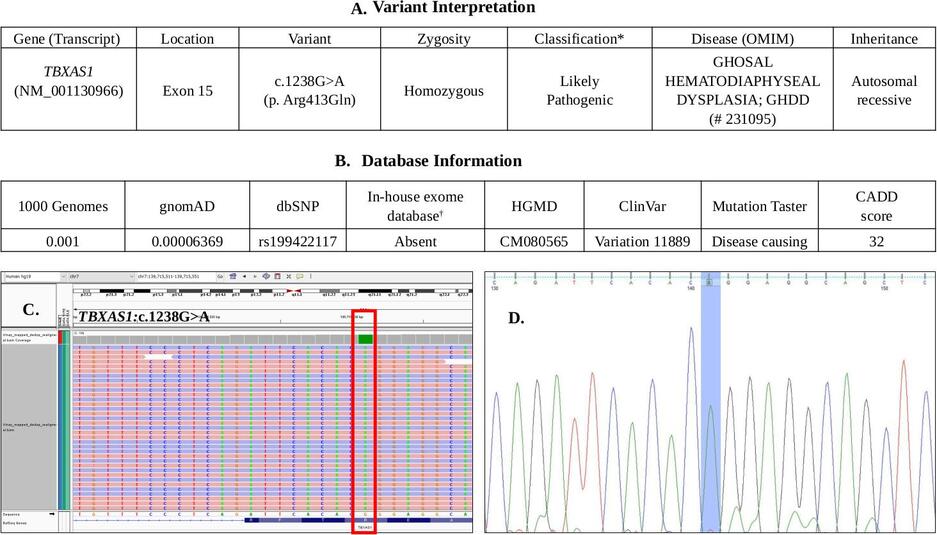
c.1238G>A (p.Arg413Gln) was identified in exon 15 of the TBXAS1 gene (transcript id ENST00000263552;
NM_001130966) (Figure 3). This variant has been previously reported in other patients of South Asian
origin with Ghosal syndrome, and is listed in HGMD (http://www.hgmd.cf.ac.uk/ac/) and ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/). The variant has minor allele frequencies of 0.001 and 0.00006 in the 1000 Genomes
(https://www.internationalgenome.org/1000-genomes-browsers/) and gnomAD (https://gnomad.broadinstitute.org/)
databases, respectively. It is classified as a ‘likely pathogenic’ variant, as per the American College of Medical Genetics
and Genomics and Association for Molecular Pathology (ACMG/AMP) guidelines (Richards et al., 2015). It has a CADD
score (https://cadd.gs.washington.edu/) of 32 and is predicted to be disease-causing/ damaging by the Mutation
Taster (http://www.mutationtaster.org/) and SIFT (https://sift.bii.a-star.edu.sg/) in-silico pathogenicity
prediction programs. The presence of the homozygous variant was validated further through targeted PCR amplification
and Sanger sequencing of exon 15 of the TBXAS1 gene in the patient’s DNA (Figure 3D), and both parents were
confirmed to be heterozygous carriers of the same. No other significant gene variants matching the phenotype were
detected.
*Evidence for classification of the variant as ‘likely pathogenic’ as per the ACMG/AMP 2015 guidelines: PM2 + PP3 + PP4 + PP5 †In-house exome database contains data from >1000 Whole exomes previously analyzed in our laboratory
Figure 3: A. Interpretation of the likely pathogenic variant identified in TBXAS1 gene B. Information
available in various population databases about the variant C. Integrative Genomics Viewer (IGV) image of
the homozygous variant identified in the TBXAS1 gene (NM_001130966: c.1238G>A) in the patient. D. Sanger
sequence chromatogram of the patient showing the same homozygous variant.
The patient was thus confirmed to have Ghosal hematodiaphyseal dysplasia. Corticosteroid therapy was
advised, but the patient was subsequently lost to follow-up and the response to therapy therefore could not be
assessed.
3 Discussion
Ghosal hematodiaphyseal dysplasia was first reported by Ghosal et al. (1988) in five children with moderate to
severe anemia and diaphyseal dysplasia. Cormier-Daire and co-workers identified the TBXAS1 gene to be
associated with this syndrome (Genevieve et al., 2008). TBXAS1 codes for the thromboxane synthase
enzyme, which is a component of the arachidonic acid cascade that produces thromboxane A2 (TXA2).
Both the enzyme and TXA2 modulate expression of osteoprotegerin and RANKL (receptor activator of
nuclear factor kappa-B ligand) which are involved in osteoclast-mediated bone resorption (Genevieve et al.,
2008).
Only 13 mutations have been reported in the TBXAS1 gene till date in the Human Gene Mutation Database
(http://www.hgmd.cf.ac.uk/ac/). The variant detected in our patient has been previously reported in other
patients of South Asian origin, including a family of Pakistani origin (Genevieve et al., 2008) and a 3 years
9 months old Indian boy with severe anemia, hepatosplenomegaly and diaphyseal dysplasia (Jeevan et
al., 2016). Our patient had chronic severe anemia, but had less severe skeletal findings and did not have
visceromegaly.
Though a rare disorder, it is relatively easy to diagnose, as it has the typical combination of skeletal dysplastic changes
(increased bone density, diaphyseal dysplasia, wide diaphyseal medullary cavities, cortical hyperostosis, metaphyseal
changes) and hematological abnormalities (anemia, thrombocytopenia, less often leucopenia, hypocellular marrow,
myelofibrosis). Molecular confirmation is essential to rule out other disorders with overlapping phenotypes particularly
Camurati-Engelmann disease.
It is important to establish the diagnosis early and accurately, because the hematological abnormalities and skeletal
changes have been found to respond very well to corticosteroid therapy and patients on low-dose long term maintenance
therapy have been reported to maintain normal hemoglobin levels, with no requirement for blood transfusions (John et
al., 2015).
This case report reiterates the importance of deep clinical phenotyping in the diagnosis and management of genetic
disorders.
References
1. Genevieve D, et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal
syndrome). Nature Genet 2008; 40: 284–286.
2. Ghosal SP, et al. Diaphyseal dysplasia associated with anemia. J Pediatr 1988;113: 49-57.
4. John RR, et al. Steroid-responsive anemia in patients of Ghosal hematodiaphyseal dysplasia: simple to
diagnose and easy to treat. J Pediatr Hematol Oncol 2015; 37: 285–289.
5. Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus
recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med 2015; 17: 405–424.