| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| Case No. | Age / Sex
CMA / MLPA Result | CNV loss
[Phenotype reported with isolated CNV loss] | CNV gain
Phenotype reported with isolated CNV gain | Dominant phenotype (CNV)
Patient’s phenotype |
| 1 | 1 year / Female Arr[hg19] 20p13p12.3(61,661-6,355,181)x3, 6q27(167,609,282-170,914,297)x1 | Het 3.3 Mb Del 6q27 16 OMIM genes [Mild GDD, Motor delay] | Het 6.2 Mb Dup 20p13-p12.3 64 OMIM genes [ID, poor motor coordination and speech, broad nasal bridge] | Dup at 20p13-p12.3 [GDD, seizures, ataxia]
|
| 2 | Fetus of 17 Weeks gestation (Sibling of case 1) Arr[hg19] 20p13p12.3(61,661-6,316,301)x3, 6q27(167,609,282-170,914,297)x1 | Het 3.3 Mb Del at 6q27 16 OMIM genes- [Mild GDD, Motor delay) | Het 6.2 Mb Dup at 20p13-p12.3 64 OMIM genes [ID, poor motor coordination, poor speech, broad nasal bridge] | Phenotype yet to evolve No dysmorphism, no gross malformations |
| 3* | 7 months / Male Arr[hg19] 22q13.31q13.33(45,143,535-51,197,766)x1, 17q25.1q25.3(74,126,271-81,041,823)x3 | Het 6 Mb Del 22q13.31-q13.33 44 OMIM genes including SHANK3 [Phelan McDermid syndrome, hypotonia, GDD, severe speech delay, prognathism, dysplastic ears, ptosis, saddle nose, normal to advanced growth, autism] (OMIM 606232) | Het 6.9 Mb Dup 17q25.1-q25.3 86 OMIM genes [GDD, FTT, distal arthrogryposis] | Blended phenotype GDD, clenched fist, bushy straight eyebrows, low set ears, triangular face, broad thumb, hypotonia |
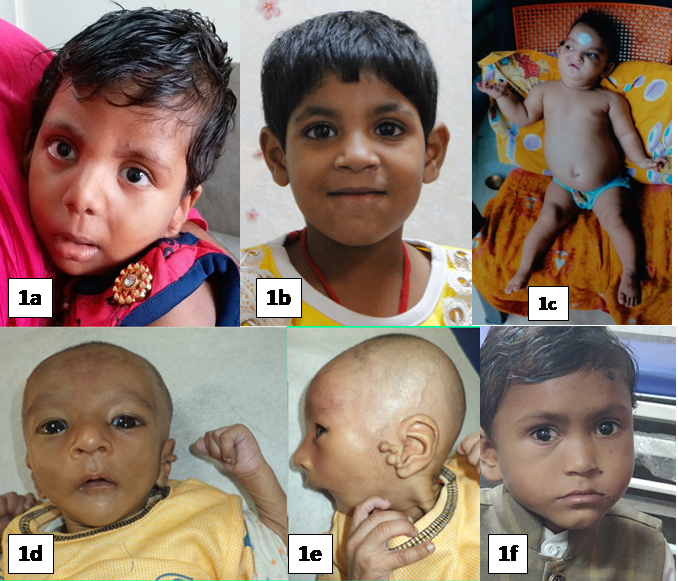
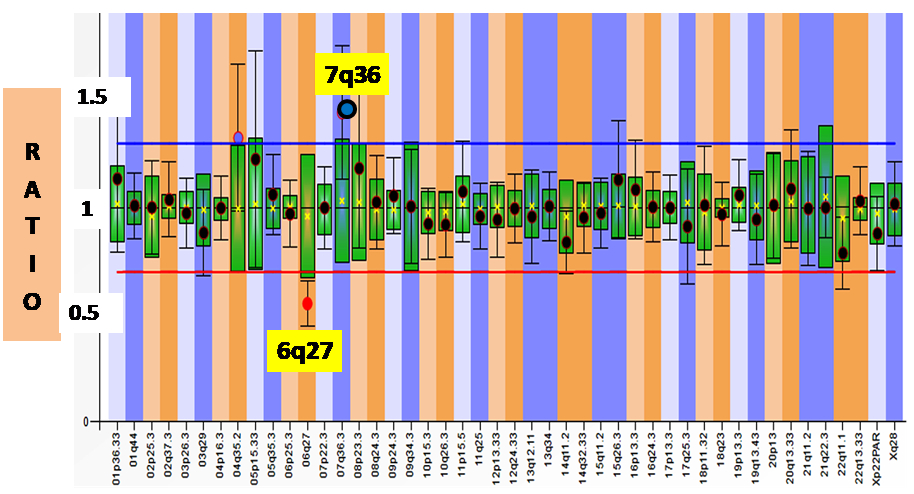
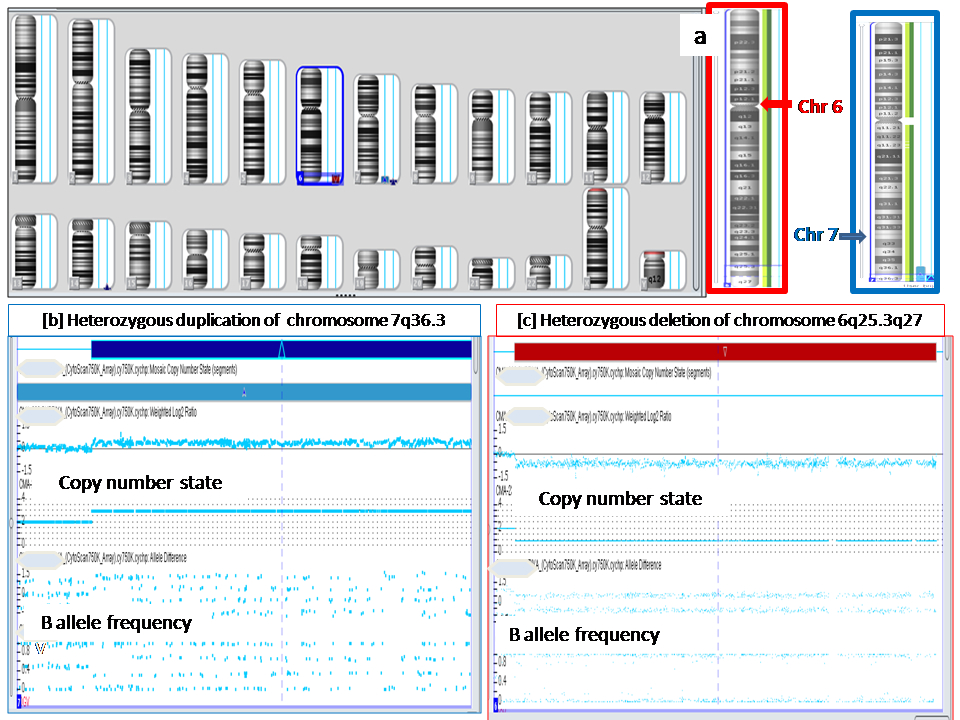
| 4* | 30 months / Female Arr[hg19] 6q25.3q27(158,628,326-170,914,297)x1, 7q36.3(155,277,221-159,119,707)x3 | Het 12.28 Mb Del at 6q25.3q27 46 OMIM genes [ID, hypotonia, epilepsy, cardiac defects, retinal abnormalities, ear anomalies, facial dysmorphisms, brain, spinal cord, and vertebrae malformations] | Het 3.8 Mb Dup at 7q36.3 12 OMIM genes. [Mild ID, macrocephaly, broad forehead, hypertelorism, muscular hypertrophy, corpus callosum agenesis] | Del 6q25.3-q27 phenotype. GDD, hypotonia, microcephaly, prominent metopic sutures, hypotelorism, micropthalmia, microcornea, smooth philtrum, and dysplastic posteriorly rotated low set ears, short neck, low posterior hairline, and bilateral talipo-equinovalgus. |
| 5 | 5 years / Female (Sibling of case 4) Heterozygous deletion at 7q36.3 and Heterozygous duplication at 6q27
| Het Del 7q36.3 #gene - VIPR2 [Speech delay, ID, short stature, holoproscencephaly, microcephaly, ptosis, sacral agenesis] | Het Dup 6q27 #gene - TBP2 [Speech delay, ID, Autistic behavior] | Predominantly social and language delay, dysmorphism
|
| 6* | 8 days / Male Arr[hg19] 5p15.33p14.3(113,576-19,902,278)x1, 7p22.3p22.2(43,376-4,156,704)x3 | Het 19.7 Mb Del 5p15.33-p14.3 54 OMIM genes [Microcephaly, round face, hypertelorism, micrognathia, epicanthal folds, low-set ears, hypotonia, and severe psychomotor and mental retardation, Cri–du-Chat syndrome] | Het 4.1 Mb Dup 7p22.3-p22.2 26 OMIM genes [ID and GDD] | Prominent Cri-du-Chat syndrome phenotype Blepharophimosis, prominent nasal bridge, left auricular tags, low set ears, retrognathia, thin tented upper lip, right 2nd and 3rd toe syndactyly |
| 7* | 30 months / Male Arr[hg19] 8p23.3p23.1(158,048-6,962,251)x1, 8q24.11q24.3(118,514,344-146,295,771)x3 | Het 6.8Mb Del 8p23.3-p23.1 15 OMIM genes [GDD, ID] | Het 27.7Mb Dup 8q24.11-q24.3 132 OMIM genes [GDD, facial dysmorphism] | GDD, facial dysmorphism, Rigidity |
| 8* | 9 months / Female Arr[hg19] 4p16.3p15.1(68,345-30,758,135)x3, 9p24.3p23(208,454-11,007,250)x1 | Het 10.8 Mb Del 9p24.3-p23 [Sex reversal syndrome] | Het 30.7 Mb Dup 4p16.3-p15.1 [GDD, minor heart defects, mild ptosis] | Blended phenotype Microcephaly, GDD, feeding difficulty, 46XY with female genitalia (sex reversal) |
| 9 | 3 months / Female Arr7q36.1q36.3 (149770238-159118443)x1, 11q24.1-25 (121769912-134926021)x3 | Het 9 Mb, loss 7q36.1 53 OMIM Genes- ASB10, SHH, XRCC2 LBW, mental retardation, GDD, facial dysmorphism, genitourinary malformations, holoprosencephaly, sacral agenesis, Currarino syndrome | Het 13Mb gain 11q24.1-25 25 OMIM genes - JAM3 Autistic behavior, Seizures, FTT, GDD, Microcephaly, IUGR, hypotonia | Blended phenotype Micro-retrognathia, FTT, agenesis of corpus callosum
|
| 10 | 5 years/ Female (Second cousin of case number 9) Arr7q36.1q36.3(149698257-159118443) x3, 11q24.1- 25(121769912-134926021] x1 | Het 13 Mb Del 11q24.1-25 25 OMIM genes including JAM3 [Jacobsen syndrome thrombocytopenia joint contractures, CHD, chorio-retinal coloboma] | Het 9 Mb gain at 7q36.1 53 OMIM Genes- ASB10, SHH, XRCC2 [Triphalangeal thumb, polysyndactyly syndrome] | Del 11q24.1-25 Phenotype GDD, facial dysmorphism, broad halluces, camptodactyly, CHD |
| 11 | 4 years / Male Heterozygous deletion at 18q23 and Heterozygous duplication at 17q13.3
| Het Del 18q23 #gene - CTDP1 [Congenital aural atresia, mild to severe developmental delay, malformation of the external ears] (OMIM 607842) | Het Dup 17q13.3 #genes - SECTM1, TBCD
| [Microcephaly, GDD, telecanthus, depressed nasal bridge with flat facial profile, low set ears, sandal gap with clinodactyly of toes, agenesis of corpus callosum, atrial septal defect] |
| 12 | Prenatal testing 16 weeks fetus Heterozygous deletion at 12p13.33 and Heterozygous duplication at 18p11.32 | Het Del 12p13.33 #gene - KDM5A Speech delay, ID, variable psychiatric manifestations | Het Dup 18p11.32, #gene - THOC1 Mild and nonspecific phenotype | History of one spontaneous abortion, history of intellectual disability in family. Mother’s Karyotype- 46, XX, t(12;18)(p13.3;p11.2) |
| 13 | Prenatal testing 11 weeks fetus (Sibling of fetus 12) Heterozygous duplication at 12p13.33 and Heterozygous deletion at 18p11.32 | Het Del 18p11.32, #gene - THOC1 ID, mild and nonspecific phenotype | Het Dup 12p13.33 #gene - KDM5A Facial dysmorphism, umbilical hernia, CNS malformations, seizures, premature ischemic stroke. |
|
| 14 | Products of conception spontaneously aborted Heterozygous deletion at 15q26.3and Heterozygous duplication at 20p13 | Het Del 15q26.3 #gene - TM2D3 IUGR, postnatal growth retardation. | Het Dup 20p13 #gene - ZCCHC3 ID, poor motor coordination and speech, broad nasal bridge | History of four recurrent spontaneous abortions.
|
| 15 | 4 year / Male Heterozygous deletion at 11q25 and Heterozygous duplication at 2q37.3
| Het Del 11q25 #gene - IGSF9B Developmental delay, short stature, CHD, thrombocytopenia, genitourinary anomalies, pyloric stenosis, and ophthalmologic anomalies | Het Dup 2q37.3 #gene - ATG4B Facial dysmorphism, hypotonia, feeding difficulties | Blended phenotype Short stature, hypertelorism, clinodactyly, fingerization of thumb with widening of wrist, bilateral undescended testes |
| 16 | 10 months / Female Heterozygous deletion at 5q35.3 and Heterozygous duplication at 19q13.43
| Del 5q35.3 #gene - GNB2L1 Developmental delay, hypotonia, FTT, postnatal short stature, CHD | Dup 19q13.43 #gene - CHMP2A Mild dysmorphic features, ID, seizures | Blended phenotype FTT, telecanthus, frontal bossing, depressed nasal bridge, smooth long philtrum, tented upper lips, spatulated nails, CHD - atrial septal defect |
| 17* | 1 month / Male Arr[hg19] 11q24.1q25(121,709,028-134,937,416)x1, 10p15.3p15.1(100,047-4,254,167)x3 Heterozygous deletion at 11q25 and Heterozygous duplication at 10p15.3 | Del 11q25 61 OMIM genes including FLI1 Developmental delay, short stature, CHD, facial dysmorphism, thrombocytopenia, genitourinary anomalies, pyloric stenosis, eye anomalies | Dup 10p15.3 33 OMIM genes Learning disability, dolichocephaly, wide sutures, frontal bossing, micro/retrognathia and renal defects
| Del 11q25 phenotype Hypertelorism, over-riding of great toes, CHD (ASD with VSD), thrombocytopenia
|
| 18 | 1 year 4 months / Female Heterozygous deletion at 4p16.3 and Heterozygous duplication at 8p23.3
| Del 4p16.3 #genes - PIGG Wolf-Hirschhorn syndrome (OMIM 194190) | Dup 8p23.3 #genes - FBXO25 GDD, ID | Del 4p16.3 phenotype GDD, facial asymmetry, bushy eyelashes, hypertelorism, broad nasal bridge, pointed low set ears, discontinuous simian crease, short stubby digits, lower limb asymmetry, pes planus, ostium secundum ASD with moderate PS with dilated RA and RV |
| 19 | Prenatal testing 16 weeks fetus Heterozygous deletion at 13q34 and Heterozygous duplication at 11q25
| Het Del 13q34 #gene - CDC16 GDD, ID, obesity, and mild facial dysmorphism. | Het Dup 11q25 #gene - IGSF9B Dysmorphic facial features, microcephaly, micrognathia, dysplastic ears, pre/postnatal growth retardation, speech delay, mental retardation, hypotonia, NTD, cardiac, vertebral, limb, urinary tract, genital anomalies. | Bad obstetric history- one first trimester spontaneous abortion and one IUD Husband’s Karyotype - 46,XY, t(11;13)(q32;q25). Previous child with 13q deletion syndrome(GDD, prenatal growth retardation, FTT, anal atresia, with recto vaginal fistula) |
| 20 | 4 years / Male Heterozygous deletion at 8p23.3 and Heterozygous duplication at 3p26.3
| Het Del 8p23.3 #gene - FBX025 GDD, ID, hypotonia, childhood onset epilepsy, autistic features | Het Dup 3p26.3 #gene - CHL1 GDD, ID, facial dysmorphism, seizures | Blended phenotype GDD, ataxia, inappropriate laughter, wooly curly hair, broad forehead, overhanging and broad nasal tip, slightly hypoplastic ala nasi, thin upper lip, MRI brain – frontal cortical atrophy. |
Note: ASD – atrial septal defect, CHD – congenital heart disease, CNV – copy number variation,
CMA – cytogenetic microarray, Del- deletion, Dup – duplication, FTT – failure to thrive,
GDD – global developmental delay, Het – heterozygous, ID – intellectual disability,
IUD – intrauterine fetal demise, PS – pulmonary stenosis, RA – right atrium, RV – right ventricle,
VSD – ventricular septal defect, IUGR – intrauterine growth retardation,
MLPA – multiplex ligation-dependent probe amplification.
* Cases where both MLPA and CMA done
# Name of the genes in the respective MLPA probes
** The phenotypic features given are from the published literature for which references are not listed.
Unbalanced chromosomal abnormalities in DSI, in which there is a net gain and loss of genetic material, often disrupt large numbers of dosage-sensitive, developmentally important genes and result in specific and complex phenotypes. Even if the sizes of the unbalanced segments are large, detection by traditional karyotyping may be difficult due to duplicated segment compensating for the size of the deleted segment. The phenotypes of DSI are combined effects of both the imbalances. In our case series, there were five prenatal samples without adequate information of phenotypes. The DSI with characteristic phenotypes seen in this series are Phelan-McDermid syndrome (case 3), Cri-du-chat syndrome (case 6), sex reversal syndrome (case 8) and Wolf-Hirschhorn syndrome (Case 18). Even in these cases, the phenotype had representation of the associated imbalance of the other chromosome. In case 3, the child had dysplastic ears as seen in Phelan-McDermid syndrome as well as distal arthrogryposis seen in duplication 17q25.1-q25.3. Five cases had blended hybrid phenotypes (Case 9,10,15,16 and 20). Case 10 had phenotype suggestive of Jacobsen syndrome (Del 11q24.1-25); however, she did not have thrombocytopenia. Most of the submicroscopic chromosomal imbalances have intellectual disability or developmental delay as features and such a non-specific phenotype was seen in six of the fifteen postnatal cases (cases 1, 4, 5, 7, 11 and 17). Predominance of phenotype of the deleted chromosomal segment was seen in some cases probably because deletion is considered more harmful than duplication. Cases of blended phenotype resulting from DSI of various chromosomal regions have been described in literature (Colangelo et al., 2018).
Phenotypic variability in combined or complex chromosomal aberrations in DSI makes it difficult to perform genotype-phenotype correlations. Hence the syndromes with characteristic phenotype may not be clinically suspected. The double segment imbalances mostly are from inherited derivative chromosomes from the parents with a balanced chromosomal rearrangement. Hence there may be history of previous spontaneous abortions, unexplained fetal losses, stillbirths or similarly affected family member. We were not able to study all the parents for carrier status by fluorescence in situ hybridization. Five of the 16 families had recurrences. The recurrence may have the same type of imbalance (cases 1 and 2) or may have the other reciprocal imbalance as was the situation in the cousins (cases 9 and 10) and the pair of sisters (cases 4 and 5). The phenotype may be similar in the recurrences as neuro-developmental disability or recurrent fetal loses. But both situations may occur in one family. Identification of these chromosomal abnormalities is of utmost importance to prevent recurrences in the family. The siblings and cousins of a carrier may be harboring the same balanced chromosomal abnormality and recurrences are expected in the extended family members. Hence, they should be offered genetic counseling and predictive testing.
Recently Iype et al. described south Indian kindred with double segment imbalance spanning five generations with t(3;4)(p26.3;p16.1), in which several individuals had either del(3p)/dup(4p) or del(4p)/dup(3p). The individuals in the family had variable phenotypes and reciprocal double segment imbalances as well. Interestingly there was no history of recurrent spontaneous abortions reported in the family (Iype et al., 2015). Nucaro et al. reported a family from Italy spanning three generations, with karyotype 46, der(3)t(3;10)(p26;p12). There was history of several miscarriages and phenotype of the patients was more likely attributable to the 10p duplication (19Mb) than to the 3p deletion (2.6 Mb) (Nucaro et al., 2008).
Rarely such a family may present with infertility in males and recurrent spontaneous abortions in female carriers (Goel & Phadke, 2011). Prenatal screening using cell-free DNA in maternal plasma or pre-implantation genetic testing are options for carriers of DSI in addition to the traditional prenatal diagnosis through fetal sampling by chorionic villus sampling or amniocentesis. Chromosomal breakpoints in the parents are important for predicting the segregation pattern and estimating the risk in the fetus. Thus, in cases of DSI, screening of the parents by karyotype or FISH is important as the risk of recurrence is high if the parent is a carrier. Many carriers of such balanced translocations may not be detectable by karyotype as the difference in the sizes of deleted and duplicated segments may not be significant and detectable by traditional karyotype.
Clinical suspicion based on phenotypes may be difficult in cases with DSI. The presence of DSI suggests a possibility of balanced chromosomal rearrangement in either of the parents. Hence identification of DSI in children, fetuses and products of conception is important. Cytogenetic microarray being a genomic technique is the best option to detect submicroscopic chromosomal imbalances. However, if it cannot be done due to cost constraints, MLPA with subtelomeric probes can be used as a substitute as it can detect DSI and thus help to identify the families at high risk of recurrence of chromosomal abnormalities.
We thank Mr Brijesh for technical support in performing Cytogenetic microarray and MLPA, and the Indian Council of Medical Research (ICMR), Government of India, New Delhi, India (grant number 63/8/2010-BMS) for funding. We are grateful to the patients enrolled in this study.
Conflict of interest
The authors declare that they have no conflict of interest.
1. Boggula VR, et al. Clinical utility of multiplex ligation-dependent probe amplification technique in identification of aetiology of unexplained mental retardation: a study in 203 Indian patients. Indian J Med Res 2014; 139: 66–75.
2. Colangelo M, et al. Case report of newborn with de novo partial trisomy 2q31.2–37.3 and monosomy 9p24.3. J Genet 2018; 97: 311-317.
3. Goel H, Phadke SR. Reciprocal balanced translocation: infertility and recurrent spontaneous abortions in a family. Andrologia 2011; 43: 75–77.
4. Iype T, et al. A large Indian family with rearrangement of chromosome 4p16 and 3p26.3 and divergent clinical presentations. BMC Med Genet 2015; 16: 104.
5. Nucaro A, et al. Familial translocation t (3; 10) (p26.3; p12.31) leading to trisomy 10p12.31→pter and monosomy 3p26.3→pter in seven members. Am J Med Genet 2008; 146A: 3242–3245.
6. Redin C, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 2017; 49: 36–45.
7. Tuteja M, et al. Double segment chromosomal imbalance due to inherited chromosomal translocation: detection by cytogenetic microarray. Indian Pediatr 2017; 54: 879–880.
| Abstract | Download PDF |