| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| X chromosome
abnormality | Phenotype | Genetic mechanism
|
| Turner syndrome (Monosomy X) | Female with dysmorphism, structural cardiac defects (one third cases), skeletal abnormalities, hearing loss (50%), hypothyroidism (10%), short stature. Milder phenotype in mosaic Turner | Non-disjunction event (meiotic or post zygotic), haploinsufficiency of SHOX gene, accelerated prenatal oocyte apoptosis |
| 47,XXX (Trisomy X) | Small proportion experience POF and may have genitourinary abnormalities. | Nondisjunction errors in meiosis I or II in oogenesis. |
| Xq deletions | Terminal deletions originating at Xq13 are associated with primary amenorrhea and absent secondary sexual development. Primary amenorrhoea is not a feature of terminal deletions arising at Xq25 or Xq26, and more distal deletions having a milder phenotype (Simpson et al.,1999). |
|
| Xp deletions | Approximately 50% of delXp11 cases show primary amenorrhea and 45% show secondary amenorrhea (Ogata et al.,1995). Deletion of only the most telomeric portion of Xp (Xp22.3 → Xpter) does not result in amenorrhea (Thomas et al., 1999). |
|
| X autosome translocations | Primary or secondary amenorrhoea. Turner stigmata if translocation occurs within the critical region of Xq13-q26 | Haploinsufficiency or disruption of critical genes in these regions, positional effect on contiguous genes or non-specific defective meiotic pairing |
| 46,XY gonadal dysgenesis | Female internal and external genitalia, minimal breast enlargement, propensity for malignant transformation of the gonads (20-30%,) | Mutations in SRY (15% of cases), SOX9, GATA4, FOG2, NR5A1, WT1, DHH, CBX2, ATRX, MAP3K1 and FGF9. Deletions encompassing DMRT1 (9p) or EMX2 (10q) Duplication of Xp21 (DAX1/NROB1) |
Perrault syndrome is characterized by 46,XX ovarian dysgenesis and bilateral prelingual onset sensorineural deafness in females. The spectrum of ovarian dysfunction extends across a continuum from mild to severe. Perrault syndrome is known to be caused by biallelic pathogenic variants in one of four genes: HARS2, HSD17B4, LARS2, or CLPP and in the majority of cases, the molecular basis is unknown. Mental retardation, ataxia, and cerebellar hypoplasia may be associated features. HSD17B4/D-bifunctional protein is a multifunctional peroxisomal enzyme involved in fatty acid β-oxidation and steroid metabolism, while LARS2 which encodes a mitochondrial leucyl-tRNA synthetase and HARS2 which codes for histidyl tRNA synthetase are mitochondrial genes.
Classical galactosemia is an inherited inborn error of galactose metabolism caused by galactose-1-phosphate uridyltransferase (GALT) deficiency and premature ovarian failure is the most common long-term complication experienced by girls and women with this condition, with more than 80% being affected despite neonatal diagnosis and careful lifelong dietary restriction of galactose. The presence of a homozygous Q188R mutation is associated with a 16-fold increased risk of POF.
A variety of genetic disorders (Table 2) have been described in which POF is a commonly occurring feature, and the genes implicated in these cases may therefore have a role in ovarian failure. In some of these conditions the onset of POF is related to gonadal insult or endocrine dysfunction resulting from the disease process, while in others the mechanism of ovarian failure remains unknown.
| Syndrome | Gene | Prominent associated findings
|
| Aromatase deficiency | CYP19A1 | Maternal virilization during pregnancy due to absence of placental aromatase |
| Ataxia telangiectasia | ATM | Cerebellar ataxia, telangiectasias, immune defects, a predisposition to malignancy, premature aging, genome instability |
| Autoimmune polyendocrine syndrome, type 1/Autoimmune polyendocrinopathy candidiasis-ectodermal dystrophy (APECED) | AIRE | Adrenal insufficiency, hypoparathyroidism, chronic mucocutaneous candidiasis <60% of patients have ovarian failure |
| Autoimmune polyendocrine syndrome, type 2 | Unknown | Adrenal insufficiency, type 1 diabetes mellitus, autoimmune thyroid disease 3-10% of APS type II patients have POF |
| Bassoe syndrome | Unknown | Muscular dystrophy and infantile cataract |
| Blepharophimosis -ptosis- epicanthus inversus syndrome (BPES) Type I | FOXL2 | Autosomal Dominant condition. Complex eyelid malformation. |
| Bloom syndrome | BLM | Premature aging, a predisposition to malignancy, genome instability |
| Cerebellar ataxia with hypergonadotropic hypogonadism | Unknown | Ataxia, sensorineural deafness with vestibular hypofunction, peripheral sensory impairment |
| Congenital adrenal hyperplasia due to 17-alpha hydroxylase deficiency | CYP17A1 | Hypertension, hypokalemic alkalosis |
| Congenital disorder of glycosylation, type 1A | PMM2 | Neonatal encephalopathy, hypotonia, psychomotor retardation, cerebellar hypoplasia, retinitis pigmentosa |
| Demirhan syndrome | BMPR1B | Severe limb malformation, genital anomalies |
| Fanconi anemia | FANCA, FACA, FA1, FA, FAA | Anemia, leucopenia, thrombocytopenia; cardiac, renal and limb malformations; dermal pigment changes |
| Fryns syndrome | PIGN & additional unknown loci | Intellectual disability, craniofacial dysmorphism |
| GAPO | ANTRX1 | Growth retardation, alopecia, pseudoanodontia, and optic atrophy |
| Leukoencephalopathy with vanishing white matter | EIF2B2, EIF2B4, EIF2B5 | Encephalopathy with leukodystrophy |
| Lipoid congenital adrenal hyperplasia | STAR | Congenital adrenal insufficiency, testis function is more severely affected than ovarian function |
| Malouf syndrome | Unknown | Cardiomyopathy |
| Marinesco-Sjogren syndrome | SIL1 | Cerebellar ataxia, congenital cataracts, retarded somatic and mental maturation |
| Mental retardation, X linked | FRAXE | Intellectual disability |
| Progressive external ophthalmoplegia with mitochondrial DNA deletions | POL G | Adult onset weakness of external eye muscles and exercise intolerance |
| Proximal symphalangism (SYM1) | NOG | Symphalangism |
| Pseudohypoparathyroidism(PHP) type Ia | GNAS | Elevated parathyroid hormone (PTH) with low/normal calcium, high thyrotropin (TSH) with normal thyroid hormone levels, growth hormone deficiency and high gonadotropins in patient with delayed puberty and skeletal abnormalities (Albright osteodystrophy) |
| Rapp-Hodgkin syndrome | TP73L | Ectodermal dysplasia, cleft lip, cleft palate |
| Werner syndrome | WRN | Premature aging, a predisposition to malignancy, genome instability |
| Woodhouse-Sakati syndrome | DCAF17 | Alopecia, diabetes mellitus, intellectual disability, extrapyramidal syndrome |
There are many X-linked genes and autosomal genes implicated in non-syndromic presentations of POF.
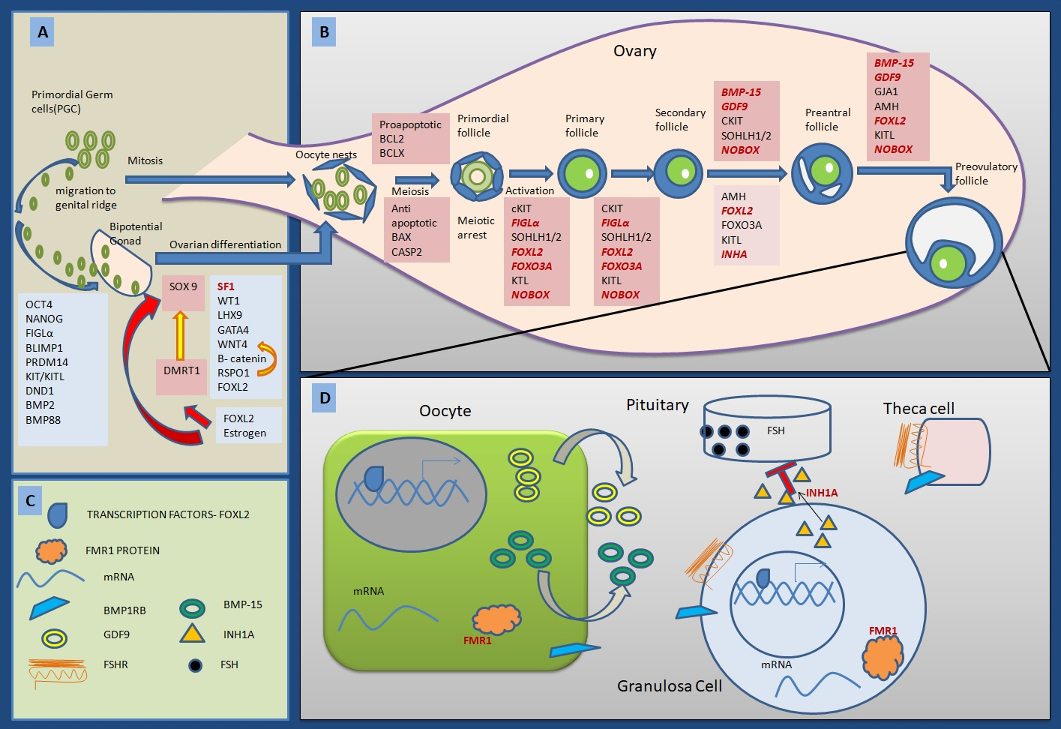
The FMR1 premutation occurs in the critical gene for Fragile X mental retardation gene located at Xq27.3. In females with POF, the risk of having a premutation allele is 3–4% when she is the only affected individual in the family, but 12–15% if a second female in the pedigree is affected with POF. The pathology is caused by expansion of the CGG repeat in the gene’s 5’ untranslated region to a premutation state of between 56 and 199 repeats which leads to an increased production of the fragile X mental retardation protein (FMRP), an RNA-binding protein which is highly expressed in germ cells of the foetal ovary Its accumulation is believed to impair the expression of genes required for oocyte development and have toxic effects leading to follicle atresia. The risk of having POF appears to increase with increasing premutation repeat size between 59 and 99. The risk plateaus or decreases for women with repeat sizes of 100. FMR1 premutations carried by women are unstable and can expand in the next generation to transmit fragile X syndrome to male offspring, especially if women have more than 100 repeats. As women with POF have an 5% chance of conceiving, these women are at risk of having a child with fragile X syndrome. As per the American College of Medical Genetics (ACMG), carrier screening of FMR1 premutation is recommended for women who are experiencing reproductive or fertility problems associated with elevated follicle stimulating hormone (FSH) levels, especially if they have: (a) a family history of premature ovarian failure, (b) a family history of fragile X syndrome, or (c) male or female relatives with undiagnosed mental retardation (Sherman et al., 2005).
Several of the isolated gene defects and their prevalence is listed below in Table 3.
| Gene | Prevalence in |
| | POF cohorts
|
| TGF-B family
| |
| BMP 15 | 1.5-12% |
| GDF9 | 1.4% |
| INHA | 0–11% |
| Gonadotropin receptors
| |
| FSH/LH resistance (FSHR and LHCGR) | 0–1% |
| Transcription factors
| |
| Nuclear Proteins | |
| NR5A1(SF1) | 1.6% |
| Oocyte specific transcription factors
| |
| NOBOX | 0–6% |
| FIGLA | 1–2% |
| Forkhead like transcription factors
| |
| FOXL2 | Rare |
| FOXO3 | 2.2% |
| Progesterone receptor membrane component 1
| |
| Progesterone receptor membrane component 1 (PGRMC1) | 1.5% |
| LHX8 | Rare |
| DNA replication/meiosis and DNA repair genes variants
| |
| DMC1, MSH4, MSH5, SPO11, STAG3, SMC1β, REC8, POF1B, HFM1, MCM8, MCM9, SYCE1, PSMC3IP, NUP107, FANCA, FANCC, FANCG | Unknown |
In rare cases, microdeletions and microduplications in known POF genes (SYCE1, CPEB1), genes involved in meiosis (PLCB1, RB1CC1, MAP4K4, RBBP8, IMMP2L, FER1L6, MEIG1) and possible candidate genes for POF and ovarian dysfunction involving DNA repair, or folliculogenesis have been identified.
MiRNAs are a class of small (18-22 nucleotides in length) noncoding RNAs which cause negative regulation of target genes by mediating post-translational gene silencing (He et al., 2004). Dicer, a pre-miRNAs processor is shown to be important for folliculogenesis, maturation of oocytes, and follicle recruitment (Murchison et al., 2007). Polymorphisms in XPO5 (Exportin), a premiRNA transporter are associated with an increased risk of POI (Rah et al., 2013). Differentially expressed miRNAs are involved in various ovarian processes and have been associated with POI (Yang et al., 2012).
In many instances, candidates genes that have been found in experimental or natural animal models showing ovarian failure have shown no variants in the corresponding human orthologue.
The lack of ovarian function leads to absence of production of ovarian hormones leading to low estradiol levels. The resulting effects represent the consequences of hypoestrogenism and also vary depending on the age at which the ovarian failure occurred. Failed development of the gonads, prenatal or prepubertal depletion of the ovarian follicles and ovarian dysfunction result in primary amenorrhoea with poor/absent secondary sexual development. The age limit for defining primary amenorrhea is 13 years of age in the absence of secondary sexual development or 15 years of age in the presence of normal secondary sexual characteristics. Secondary amenorrhea (as absence of menstruation for three normal menstrual cycle or four months period) and well developed secondary sexual characteristics are features of post-pubertal events. A preceding history of infertility, recurrent pregnancy loss or irregular cycles is usually elicitable in such cases. Other symptoms of POF are the typical manifestations of climacterium such as palpitations, heat intolerance, flushes, night sweats, irritability, anxiety, depression, sleep disturbance, decreased libido, hair coarseness, vaginal dryness, fatigue. These symptoms are uncommon among women with primary amenorrhea who never received estrogen. Over 75% of women with POI will have at least menopausal intermittent symptoms including hot flushes, night sweats, and emotional lability. Moderate hirsutism may be seen due to the action of androgens originating from the adrenal glands. In addition to these common manifestations, some have additional specific features associated with specific syndromes or etiologies (see Table 2).
The clinical assessment of a woman with premature ovarian failure is aimed at finding etiological clues. These include determining the age of onset of amenorrhea, the sexual maturity rating (SMR), anthropometry, dysmorphological assessment, systemic examination for associated features such as cardiac abnormalities or signs of endocrinological disturbances and examination of the external genitalia. A comprehensive three generation pedigree for history of familial POF and for members affected with Fragile X syndrome or ataxia, and maternal menarcheal and menopausal age may provide vital information. A sonographic evaluation of the pelvis helps to delineate the pelvic anatomy, presence of female internal genital organs, and uterine and ovarian morphology. Relevant imaging and laboratory tests may then be undertaken to further aid in establishing the diagnosis and optimize patient management. In every woman of reproductive age with amenorrhea pregnancy should be ruled out. Serum FSH levels of greater than 40 mIU/ml are diagnostic of POF and this is confirmed with a repeat value after four weeks. About half of the cases of primary amenorrhea are due to ovarian dysgenesis, which is revealed by the finding of streak ovaries accompanied by uterus hypoplasia at ultrasound. In the other patients, follicles (<10 mm) may be found on histological evaluation, such as in the case of FSHR gene mutations.
A routine karyotype is performed in all cases with premature ovarian failure regardless of the age of onset. Besides detecting X chromosome abnormalities, a karyotype will help identify any Y chromosome material which necessitates gonadectomy due to the associated risk of gonadal tumors. The ACMG 2005 guidelines and the American College of Obstetrics and Gynaecology (ACOG) 2010 committee opinion recommend Fragile X premutation screening in women with unexplained premature ovarian insufficiency (Sherman et al.,2005; ACOG committee opinion., 2006). All identified premutation carriers should be counseled regarding the risk to their offspring of inheriting an expanded full-mutation Fragile X allele and also the importance of cascade screening of at-risk female relatives. Currently there are no recommendations for the routine testing of other candidate genes associated with POF in cases with a normal karyotype. Testing of specific disease-associated genes can be considered if a particular syndrome is suspected.
Strategies to identify POF candidate genes have included studies in animal models, study of X chromosome deletions and X-autosome translocations, linkage analysis, comparative genomic hybridization (CGH) array, genome-wide association studies (GWAS), and recently, next generation sequencing (NGS) based approaches. Of these, NGS has several advantages and is a powerful diagnostic tool. The mitochondrial gene linked to Perrault syndrome LARS2, which codes for a mitochondrial leucyl-tRNA synthetase, was identified by exome sequencing in two POF families (Pierce et al., 2013). Studies involving whole exome sequencing(WES) have identified pathogenic variants in genes implicated in DNA repair and genomic stability such as stromal antigen 3 (STAG3), synaptonemal complex central element 1 (SYCE1), minichromosome maintenance complex component 8 and 9 (MCM8, MCM9) and ATP-dependent DNA helicase homolog (HFM1) genes in consanguineous families with non-syndromic POF (Carburet et al., 2014; Wood-Trageser et al., 2014; Al Asiri et al., 2015; Wang et al., 2014; de Vries et al., 2014). A multigene panel study by Fonseca et al. in 12 unrelated women with POF identified two plausible candidate genes in POF, namely ADAMTS19 and BMP receptor 2 (BMPR2) genes with POF pathogenesis (Fonseca et al., 2015). Candidate gene discoveries are important to enhance the knowledge of the underlying molecular mechanisms of POF and thereby increase the prospects of development of definitive treatment.
Recently, the concept of POF being a purely monogenic disorder has been questioned with digenic findings in several cases and the synergistic effects of several mutations have been suggested to underlie the POI phenotype (Bouilly et al., 2016). Research in POF besides being directed towards identifying causative gene defects, involves exploring therapeutic options to restore ovarian function. In vitro activation of dormant primordial germ cells and grafting are being studied as a potential infertility therapy for POF patients who have residual follicles (Kawamura et al., 2013; Suzuki et al., 2015). The online Ovarian Kaleidoscope Database (http://ovary.stanford.edu) provides information regarding the biological function, expression pattern and regulation of genes expressed in the ovary. It also contains information on gene sequences and is a useful resource of knowledge on ovarian genetics.
Currently there are no proven predictive tests or biomarkers to identify women who will develop POF, unless a mutation known to be related to POI is detected, and there are no established POI preventing measures. The development of multigene predictive panels may enable the identification of women at risk for early menopause or premature ovarian failure. The availability of target therapies is the ultimate goal for the immense ongoing research in unravelling the intricacies of the ovarian function.
1. ACOG Committee on Gynecologic Practice. ACOG committee opinion no. 357: Primary and preventive care: Periodic assessments. Obstet Gynecol 2006;108:1615-1622.
2. Al Asiri S, et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J Clin Invest 2015;125: 258-262.
3. Bouilly J, et al. Identification of Multiple Gene Mutations Accounts for a new Genetic Architecture of Primary Ovarian Insufficiency. J Clin Endocrinol Metab 2016;101: 4541-4550.
4. Caburet S, et al. Mutant Cohesin in Premature Ovarian Failure. N Engl J Med 2014; 370: 943-949.
5. Coulam CB, et al. Premature gonadal failure. Fertil Steril 1982; 38: 645-655.
6. Coulam CB, et al. Incidence of premature ovarian failure. Obstet Gynecol 1986; 67: 604-606.
7. Dalpr L, et al. Cytogenetics of premature ovarian failure: An investigation on 269 affected women. J Biomed Biotechnol 2011; 2011: 370195.
8. de Vries L, et al.Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J Clin Endocrinol Metab 2014; 99: E2129-132.
9. Eggers S, et al. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol 2014; 10: 673-683.
10. Fonseca DJ, et al. Next generation sequencing in women affected by nonsyndromic premature ovarian failure displays new potential causative genes and mutations. Fertil Steril 2015; 104: 154-162.
11. Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update. 2005;11: 391-410.
12. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522-531.
13. Jiao X, et al. Cytogenetic analysis of 531 Chinese women with premature ovarian failure. Hum Reprod 2012; 27: 2201-2207.
14. Kalantari H, et al. Cytogenetic analysis of 179 Iranian women with premature ovarian failure. Gynecol Endocrinol 2013; 29: 588-591.
15. Kawamura K, et al. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proc Natl Acad Sci 2013; 110:17474-17479.
16. Murchison EP, et al. Critical roles for Dicer in the female germline. Genes Dev. 2007; 21: 682-693.
17. Ogata T, Matsuo N. Turner syndrome and female sex chromosome aberrations: deduction of the principal factors involved in the development of clinical features. Hum Genet 1995; 95: 607-629.
18. Persani L, et al. Genes involved in human premature ovarian failure. J Mol Endocrinol 2010; 45: 257-279.
19. Pierce SB, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 2013; 92: 614-620.
20. Qin Y, et al. Genetics of primary ovarian insufficiency: New developments and opportunities. Hum Reprod Update 2015; 21: 787-808.
21. Rah H, et al. Association of polymorphisms in microRNA machinery genes (DROSHA, DICER1, RAN, and XPO5) with risk of idiopathic primary ovarian insufficiency in Korean women. Menopause 2013; 20:1067-1073.
22. Sherman S, et al. Fragile X syndrome: Diagnostic and carrier testing. Genet Med 2005; 7: 584-587.
23. Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet 1999; 89:186-200.
24. Suzuki N, et al. Successful fertility preservation following ovarian tissue vitrification in patients with primary ovarian insufficiency. Hum Reprod 2015; 30: 608-615.
25. Therman E, et al. The critical region on the human Xq. Hum Genet 1990; 85: 455-461.
26. Toniolo D, Rizzolio F. X chromosome and ovarian failure. Semin Reprod Med 2007; 25: 264-271.
27. Thomas NS, et al. Xp deletions associated with autism in three females. Hum Genet 1999;104: 43-48.
28. Van Kasteren YM, et al. Familial idiopathic premature ovarian failure: An overrated and underestimated genetic disease? Hum Reprod 1999; 14: 2455-2459.
29. Wang J, et al. Mutations in HFM1 in Recessive Primary Ovarian Insufficiency. N Engl J Med 2014; 370: 972-974.
30. Welt CK. Primary ovarian insufficiency: A more accurate term for premature ovarian failure. Clin Endocrinol (Oxf) 2008; 68: 499-509.
31. Wood-Trageser MA, et al. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am J Hum Genet 2014; 95: 754-762.
32. Yang X, et al. Differentially expressed plasma microRNAs in premature ovarian failure patients and the potential regulatory function of mir-23a in granulosa cell apoptosis. Reproduction 2012;144: 235-244.
| Abstract | Download PDF |