The Utility and Futility of Targeted Next Generation Sequencing for Carrier Detection in At Risk Couples
Sunita Bijarnia-Mahay 1*, Deepti Gupta 1, V L Ramprasad 2, Sakthivel Murugan 2, Renu Saxena 1, Sudha Kohli 1, Seiji Yamaguchi 3, Yosuke Shigematsu 4and I C Verma 1 1Institute of Medical Genetics & Genomics, Sir Ganga Ram Hospital, New Delhi, India 2Medgenome Labs Pvt. Ltd, Bommasandra, Hosur Road, Bangalore, Karnataka, India 3Department of Pediatrics, Shimane University School of Medicine, Izumo Shimane, Japan 4Department of Pediatrics, University of Fukui, Fukui, Japan Email:bijarnia@gmail.com
1 Abstract
Next generation sequencing has changed the approach to genetic diagnosis and testing in recent times. The days have
arrived when a molecular genetic diagnosis can be attempted even without the availability of the proband or affected
person. However this requires additional strong evidence of diagnosis in the proband, such as biochemical or radiological
hallmarks. Needless to say, this attempt should always be made during counseling to make the family aware
of the fallacies and limitation involved due to non-availability of the sample of the proband. Since many
recessive disorders are either fatal or severely debilitating and burdensome, with no easy treatment, the NGS
technology has provided a much needed relief in terms of testing and prevention in family by enabling
prenatal diagnosis, at least in a few cases. Encouraging results have been shown upon testing of the proband
directly. However, the final word remains to be said on complete reliability of the method for carrier testing,
performed without availability of the proband. We present two similar cases, of methyl malonic acidemia, where
use of NGS resulted in contrasting outcomes after genetic testing for carrier status, thus highlighting the
utility and the futility of the test in certain situations. While a correct identification of mutations in the
first couple (in the MMAB gene) resulted in successful prenatal diagnosis and prevention of recurrence, an
inability to identify the disease and mutation resulted in birth of an affected baby with MMA in the second
family.
2 Introduction
Next generation sequencing (NGS) technology has revolutionized genetic testing by providing an easy, and relatively quick
and accurate solution to the ‘undiagnosed genetic disorders’ group (Katsanis et al., 2013). It has landed in a big way in
India, especially for enabling prenatal diagnosis, since majority of families, at least in our set up, present with the hope
of prevention of recurrence of their burdensome disease, in their ongoing or future pregnancies (Puri et
al., 2016). Therefore, the main purpose of genetic testing and diagnosis in the proband or family member
is essentially to be able to make a confirmed molecular diagnosis, so as to enable an accurate prenatal
diagnosis. NGS in parents of an affected child, where the index case or proband is unavailable, is not that
straightforward. In specific situations where a phenotype is clearly defined, and there is an additional strong
evidence available (such as biochemical or radiological), carrier testing may be considered. Inborn errors
of metabolism, where the diagnosis is usually achieved using biochemical metabolic testing, is one good
example. However, there remain a lot of uncertainties if this approach is applied, as illustrated by the two
cases described here. The two cases chosen had a similar background history and ultimately proved to have
the same diagnosis, but with vastly different consequences after using the same technology. Both cases
had a diagnosis of methylmalonic acidemia, which is the commonest organic acidemia in India (Verma
et al., 2005). Methylmalonic acidemia is an autosomal recessive IEM of energy production. The clinical
presentation is very variable, from neonatal fatal presentation if unrecognized, to mild intermittent illness. The
disorder is manageable if recognized in time, and appropriate treatment is instituted (Baumgartner et al.,
2014).
3 Case 1
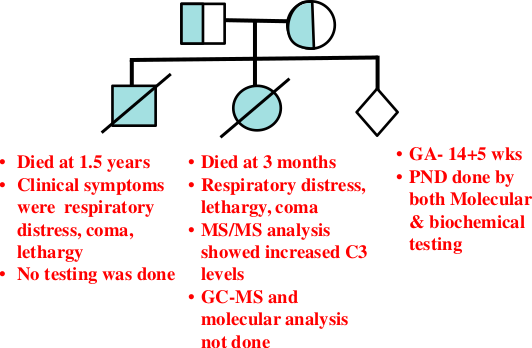
A non-consanguineous couple, from Goa, presented to us in the pre-pregnancy planning stage for genetic counseling. The
family had lost two children at 1.5 years and 3 months of age, with episodes of respiratory distress, lethargy and coma
with no clear diagnosis (Figure 1). Tandem mass spectrometry (MS/MS) analysis in the second baby had shown increased
C3 metabolite, suggesting the possibility of organic acidemia. Due to non-availability of the proband’s DNA, the
couple was tested, sequentially. The wife’s DNA was tested for multigene panel of organic acidemias using
NGS. A heterozygous novel mutation, c.2T>G, in the initiation codon, was detected in the MMAB gene.
In-silico analysis predicted pathogenicity.. The variation, c.2T>G was considered pathogenic as the initiation
codon is lost. The highly conserved ‘T’ of ATG is substituted by ‘G’. Due to this change, translational
process does not start at the expected position and requires an alternative downstream initiation codon
(Wolf et al., 2011). The next in-frame methionine codon is present 600bp downstream and would comprise
approximately 50 amino acids. If it is used it is expected to lead to a truncated protein without the N-terminal
domain and hence is unlikely to be functional. The MutationTaster software predicted this mutation to be
disease-causing.
Figure 1: Case 1: Pedigree with clinical details of the two affected children.
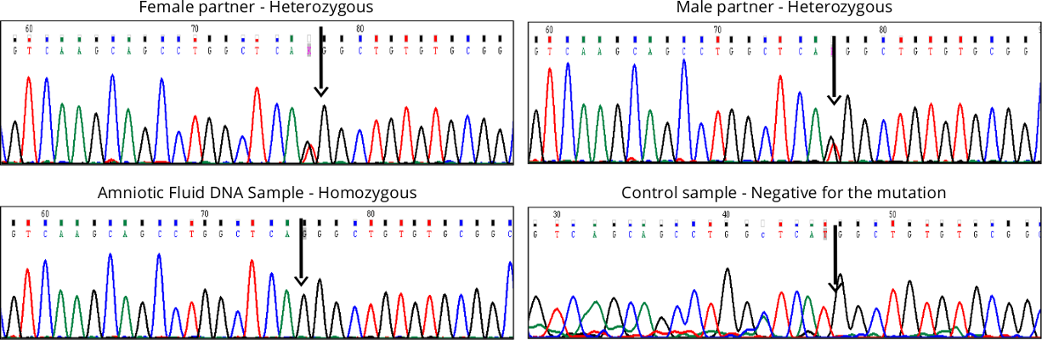
After validating the mutation using Sanger sequencing, the husband’s DNA was sequenced and noted to carry the
same mutation (Figure 2). Prenatal diagnosis was performed in the subsequent pregnancy, using amniotic fluid for
metabolite testing through Gas Chromatography Mass Spectrometry (GC-MS) as well as targeted mutation analysis. The
biochemical analysis on AF showed that the fetus was affected, with elevation of methylmalonate in the amniotic fluid.
The same was also confirmed by mutation analysis (homozygous c.2T>G) (Figure 2). The pregnancy was terminated after
counselling.
Figure 2: Molecular studies in family (Case 1) showing presence of mutation, c.2T>G in the MMAB gene in
heterozygous state in the couple, and homozygous state in the fetus (amniocyte DNA), and normal sequence in
a control sample.
Targeted panel sequencing thus proved useful in this family as it paved the way for prenatal diagnosis and prevention
of this burdensome disorder in the family. The results were convincing as there was additional biochemical evidence of
disease in the fetus.
4 Case 2
A non-consanguineous couple from North India presented in the pre-pregnancy period for genetic counseling. The couple
had lost a previous baby in the neonatal period. The baby girl was born at term, uneventfully. She was
admitted on day 7 of life in NICU with history of lethargy and poor feeding. Baby was in shock at the
time of admission, with tachypnoea and severe metabolic acidosis. She was treated on the lines of late
onset sepsis, but went into refractory shock on day 2 of admission and succumbed. Routine hematology,
biochemistry, liver and renal functions were normal with only mild derangement in the coagulation profile and
no evidence of sepsis. Metabolic test (tandem mass spectrometry, TMS) was done which showed elevated
propionylcarnitine (C3) metabolite on dried blood spot (11 �mol/L, normal<6). There was also increase
in a few amino acids (leucine, ornithine, phenylalanine, tyrosine and valine) which suggested secondary
metabolic derangement owing to sick state and liver dysfunction. Baby died on day 10 of life, 48 hours after
admission.
Considering the presentation of the baby, a heightened suspicion of an organic acidemia was made (severe metabolic
acidosis, raised C3 on TMS, no evidence of sepsis). The mother of the baby was tested for carrier status, using the
targeted organic acidemia panel by next generation sequencing. A panel of genes causing organic acidemia was tested. No
specific mutation, correlating with the disease in the baby was noted. However, she was noted to be heterozygous for
a novel, possibly significant variant (c.278InsA in ETFB gene). Homozygous or compound heterozygous
mutations in the ETFB gene result in Multiple Acyl CoA dehydrogenase (MADD) deficiency, also known as
Glutaric Aciduria type 2. The neonatal form of MADD is usually fatal and characterized by severe nonketotic
hypoglycemia, metabolic acidosis, multisystem involvement, and excretion of large amounts of fatty acid- and amino
acid-derived metabolites. In view of a positive clinical correlation, this mutation was considered significant and
led to testing of the husband. Again, NGS for organic acidemia genes was performed and he was noted
to be negative for the same mutation and also negative for any other pathogenic variation in the ETFB
gene.
The couple was then counseled regarding the non-feasibility of prenatal diagnosis in their subsequent pregnancy owing
to failure of detection of their carrier status using the NGS test. The couple went ahead with pregnancy, with routine
monitoring and delivered a healthy boy at term, weighing 2.5 kgs. He was kept under observation for 4 days in the
hospital and discharged on breast feeds. Newborn screening was performed on day 3 of life using TMS which
detected elevated levels of C3 (5.09 nmol/ml, normal <3.5), with normal levels of amino acids. Further testing
showed elevated methylmalonic acid (24.2 �M; ref.<1) on DBS. Urine GC-MS analysis revealed elevation of
methylmalonate, 3-hydroxypropionate and methylcitrate, consistent with the diagnosis of Methyl malonic
acidemia. Serum homocysteine level was not elevated. The baby was started on the treatment regimen: Inj.
Hydroxocobalamin 1 mg per day intramuscularly, oral carnitine and vitamin supplements and continued
on breast feeds. A repeat GC-MS after few weeks of vitamin B12 therapy revealed a marked reduction
in the peak of methylmalonate in the urine sample, suggesting B12-responsiveness. The baby is now 16
months old and doing well, gaining milestones appropriately, with no major decompensations requiring
hospitalization.
Molecular testing in the baby, using the NGS- based organic acidemia panel did not reveal any pathogenic
variation. However, note was made of one large contiguous region corresponding to exon 5 in the MMAA gene
(chr4:146572210-146572303) that was not covered in the sequence data of the sample, which is usually covered by the test.
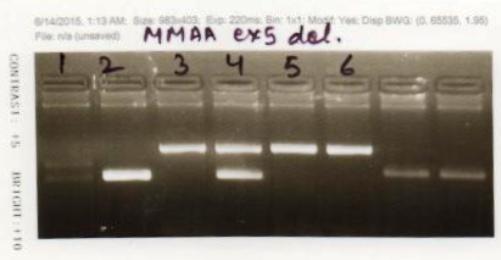
This suggested possibility of a homozygous deletion of exon 5 in the MMAA gene. The same was then tested using PCR
using specific primers for exon 5 of the MMAA gene (Figure 3) , which confirmed the diagnosis of MMA in the
child.
Figure 3: Molecular studies for detection of homozygous deletion of exon 5 in MMAA gene in the proband.
PCR analysis was performed using specific primers for exon 5 (Lanes 1 & 2) and also multiplexing with another
PCR for amplification of a fragment of HEXA gene (Lanes 3 & 4) in patient and control DNA. PCR analysis of
only HEXA gene fragment is also shown (Lanes 5 & 6). Lanes 1, 3 and 5 contain patient’s DNA and Lanes 2, 4
& 6 contain control DNA. Lane 1 with patient’s DNA shows no band, thus no amplification of exon 5 after PCR
using exon 5 specific primers; Lane 2 shows positive band of exon 5 in a control sample; Lane 3 shows positive
band for the HEXA fragment and no band for the exon 5 MMAA gene fragment in patient‘s DNA; Lane 4 shows
presence of both HEXA and MMAA fragments in control DNA; Lane 5 & 6 show positive bands for HEXA in the
patient’s DNA and control DNA respectively.
Homozygous deletion of exon 5 in the proband was confirmed by multiplexing exon 5 of MMAA gene with another
unrelated fragment (exon 7 of HEXA gene), where exon 7 HEXA served as a control fragment. In this multiplex PCR the
proband’s DNA amplified for exon 7 HEXA while exon 5 of MMAB gene did not amplify, proving deletion of exon 5. The
heterozygous deletion in parents could not be confirmed by the same PCR as their DNA amplified for exon 5 of the
MMAB gene.
5 Discussion
Methyl Malonic Aciduria (MMA) is a common organic aciduria observed in the Indian population (Verma et al., 2005).
Although diagnosis of MMA is achieved by biochemical testing based on abnormal metabolites on MS/MS on dried blood
spots and GC/MS in patient’s urine sample, molecular diagnosis is still required to provide confirmation of
the exact type of MMA and to enable an accurate prenatal diagnosis for subsequent pregnancies. Next
generation sequencing is a powerful technology benefitting particularly the average Indian family who wish to
undergo prenatal diagnosis to prevent the burden of a genetic disorder in the family. While the technology has
been used very successfully in making an accurate genetic diagnosis in probands (Reid; 2016), this has
not been evaluated in the ‘at risk’ family members who may be carriers. Various levels of testing are now
possible using NGS varying from a targeted panel of genes, to whole exome or whole genome sequencing
(Katsanis et al., 2013). We utilized the targeted panel of genes to identify carrier status in two couples
presenting with a similar history. After going through the similar process of carrier testing, the outcome
was starkly different. While next generation sequencing (NGS) enabled prenatal diagnosis by molecular
analysis in the first family, with successful prevention of recurrence, this was not possible in the second
family. Thus, clinicians should be careful in counseling the families, especially couples who approach for
testing, especially in the absence of a proband, about the lack of complete reliability on the NGS for genetic
testing.
The two cases also illustrate the finding which is increasingly being noted in other Indian studies, which is the presence
of homozygosity even in absence of any consanguinity (Gupta et al., 2016). This is very depictive of the unique system of
marriages in our country where small pockets of homogeneity are created within the heterogeneous population, because of
a caste or community barrier.
References
1. Baumgartner MR, et al. Proposed guidelines for the diagnosis and management of methylmalonic and
propionic acidemia. Orphanet J Rare Dis 2014; 9:130.
2. Gupta D, et al. Seventeen novel mutations in PCCA and PCCB Genes in Indian propionic acidemia patients,
and their outcomes. Genet Test Mol Biomarkers 2016; 20: 373-382.
3. Katsanis SH and Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet
2013; 14: 415-426.
4. Puri RD, et al. Genetic approach to diagnosis of intellectual disability. Indian J Pediatr 2016; 83:1141-1149.
5. Reid ES, et al. Advantages and pitfalls of an extended gene panel for investigating complex neurometabolic
phenotypes. Brain 2016 Sep 6. pii: aww221. [Epub ahead of print]. doi: 10.1093/brain/aww221.
6. Verma IC, et al. Screening for inborn errors of metabolism. J Neonatol 2005; 19:107-116.
7. Wolf A., et al. Single base-pair substitutions at the translation initiation sites of human genes as a cause
of inherited disease. Hum. Mutat 2011, 32: 1137e1143.