| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
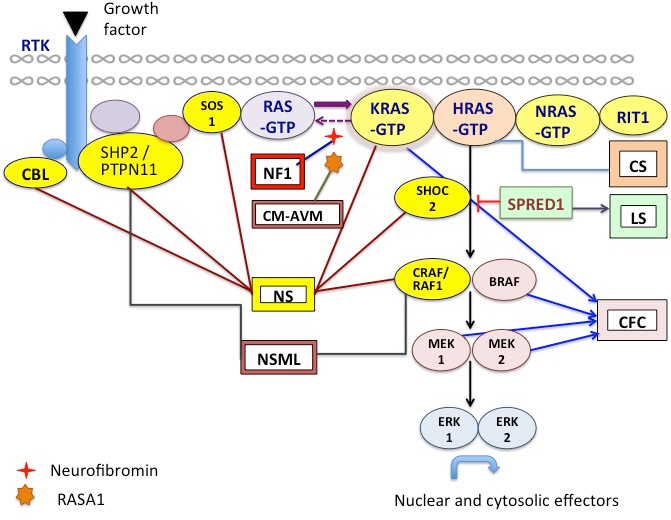
GeNeViSTA
| Syndrome |
RAS/MAPK
pathway
gene |
Proportion
of disease
attributed
to this
gene
|
| Neurofibromatosis 1 | NF1 | >95% |
| Noonan Syndrome (NS) | PTPN11 | 50% |
|
| SOS1 | 10%-13% |
|
| RAF1 | 3%-17% |
|
| KRAS | <5% |
|
| NRAS | 4 individuals to date |
|
| BRAF | <2% |
|
| MAP2K1 | <2% |
|
| SHOC2 | 2% |
|
| CBL |
|
| Noonan syndrome with multiple lentigines | PTPN11 | 90% |
|
| RAF1 | <5% |
|
| BRAF | 2 individuals |
|
| MAP2K1 | 1 individual |
| Capillary malformation-arteriovenous malformation | RASA1 | >70% |
| Costello syndrome | HRAS | 80%-90% |
| Cardio-facio-cutaneous syndrome | BRAF | 75% |
|
| MAP2K1 | 25% |
|
| MAP2K2 |
|
|
| KRAS | <2%-3% |
| Legius syndrome | SPRED1 | 98% |
| Novel genes in NS and NS like syndromes (No. of families reported) | RIT1,
RRAS, RASA2, A2ML1,
SOS2, LZTR1 (Bezniakow
N, et al., 2014; Korf et
al., 2015; Aoki et al., 2016;
Allanson & Roberts, 2011)
| |
We discuss here a few of the interesting cases that presented to the genetic clinic at our hospital from 2012 through December 2015, with characteristic features of Noonan and other RASopathies. Neurofibromatosis type 1 is not included in this series except one case of NF1 with developmental delay and craniofacial dysmorphism reminiscent of NS. A total of 20 patients including 13 patients with a provisional diagnosis of Noonan syndrome (NS), 5 patients with a diagnosis of Cardiofaciocutaneous syndrome (CFC), 1 patient with suspected NF - Noonan syndrome and 1 patient of Costello syndrome were seen during this time. The age of presentation varied from 3 months to 22 years and one fetus was evaluated after termination of the pregnancy at 18 weeks gestation. The clinical data of the patients is presented in the Table 2.
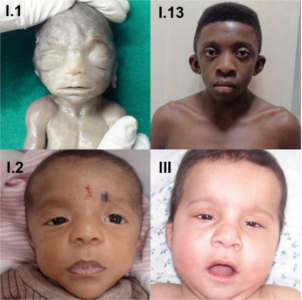
The most consistently found feature in all patients was the characteristic facial dysmorphism - low set and posteriorly rotated ears, broad nasal bridge, hypertelorism and downslanting palpebral fissures seen in 17/20 (85% of patients). Additionally ptosis was noted in 12/20 (60% of patients) (Fig 2A). In patients with clinical suspicion of cardiofaciocutaneous syndrome (5/20 patients), the face was broader and coarse looking with sparse, thick, curly wooly hair (Fig 2B). In our one patient of Costello syndrome (Table 2-IV) from Nigeria, the facies were much more coarse with thick, fleshy earlobes, full nasal tip and thick lips (Fig 2C; the mother’s photo given for comparison). The facial features associated with NS and related disorders vary considerably with age, being most striking in the neonatal period and childhood. Since the presentation can be mild and the typical facies recede with age, the diagnosis may be overlooked. Hence, the facial dysmorphism should be carefully noted at the initial visit as “gestalt” assessment is the commonest diagnostic tool for disorders of the RAS-MAPK pathway.
|
Patient
ID | Age
at
presentation
/Sex | Antenatal |
Development
/Intellect |
Stature | Heart
disease | Facial
features | Skeletal | Others/
Mutation
|
| I.1 | 18 weeks fetus / M | Bilateral hypoplastic kidneys with oligohydramnios. Echocardiography- Pulmonary stenosis. | - | Crown rump length: 18 cms (18-19 weeks) | Pulmonary stenosis | Telecanthus, broad nose, low set ears, downslanting palpebral fissures | Normal radiographs | - |
| I.2 | 3 months / M | NT-5.6 mm, Femur - 5th centile for gestation, Antenatal Karyotype- 46,–, Normal | Appropriate | 59 cms (50th centile) | not present | Sparse eyebrows, downslanting eyes, triangular chin, low set ears, high arched palate, bulbous tip of nose | Right CTEV, limitation of elbow joint extension, right clinodactyly. |
|
| I.3 | 5.5 months / M | Antenatal data not available. | Appropriate | 55.6 cms (- 4 SD) | Hypertrophic cardiomyopathy | Downslanting eyes, Bulbous nasal tip | Short neck, shield like chest, wide spaced nipples | - |
| I.4 | 5.5 months / M | Antenatal period- uneventful | Global development delay | 56 cms (-4SD) | Atrioventricular canal defects | No dysmorphic features | - | Juvenile myelomonocytic leukemia, feeding difficulty & vomiting, retrocollis failure to thrive, loose skin, scant hair. Heterozygous mutation (c.218C>T, p.Thr73Ile) in PTPN11 gene |
| I.5 | 10 month / M | Antenatal data not available | Appropriate | 63 cms (-3 SD) | ASD with severe valvular pulmonary stenosis | Downslanting eyes | - | Feeding difficulty & vomiting, bilateral undescended testes |
| I.6 | 1 year / M | Antenatal data not available | Appropriate | 70 cms (-2 SD) | Severe valvular pulmonary stenosis with RVH | Facial asymmetry, left ptosis, hypertelorism, epicanthic folds | Bilateral 2nd-3rd toe syndactyly, left Simian crease | Micropenis |
| I.7 | 4 years / M | Antenatal data not available | Mild development delay | 90 cms (-2 to -3 SD) | Atrial septal defect | Downslanting eyes, hypertelorism | - | Undescended testes, hypospadias |
| I.8 | 4 years / M | Antenatal data not available | Normal | 91 cms (-3 to -2 SD) at diagnosis | Atrialseptal defect | Ptosis, broad nasal bridge | - | On Growth hormone therapy |
| I.9 | 4.5 years / M | Antenatal data not available | Normal | 88 cms (-5 to -4 SD) at diagnosis | Supravalvular tethering of pulmonary valve | Mild ptosis, depressed nasal bridge | Winging of scapula, pectus excavatum, short neck, limitation of elbow extension | On Growth hormone therapy |
| I.10 | 7 years / M | Antenatal data not available | Development delay present | 100.5 cms (-4 SD) | Soft systolic murmur, recurrent respiratory infections. Echo - Not done. | Downslant palpebral fissures, low set ears, small philtrum, teeth pigmentation | Short neck, small hands and feet | CT head- Hydrocephalus Heterozygous mutation (c.2536G>A, p.Glu346Lys) in exon 16 of SOS1 gene |
| I.11 | 10 years 8 month / F | Antenatal data not available | Mild ID | 111 cms (-5 SD) | Echocadiography - Normal | Ptosis | Short neck, pectus carinatum |
|
| I.12 | 14 years / F | Antenatal data not available | Mild ID | 135 cms (-3 SD) | Pulmonic stenosis | Epicanthal folds, hypertelorism, low nasal bridge, downward eye slant, low set ears | - | Lentigines |
| I.13 | 22 years / M | Antenatal data not available | Mild ID | 158 cms | CHD- Unspecified | Broad nasal bridge, maxillary hypoplasia, small curved eyelashes, preauricular sinus | Bilateral pedal edema, broad laterally deviated toes | - |
| II.1 | 1 year / F | Unilateral hydronephrosis | Severe development delay | 68 cms (-3 to -2 SD) | Pulmonary stenosis | Depressed nasal root, wide base of nose, bulbous tip | - | Feeding difficulty with GERD |
| II.2 | 3 years / F | Polyhydramnios | Mild global development delay | 87 cms (3- 50th centile) | Left ventricle hypertrophy, left ventricle dysfunction, mild mitral regurgitation | Coarse facies, downslant palpebral fissures, low set ears, broad forehead, hypertelorism, strabismus | Short broad thumbs, fingers and toes, widely spaced nipples | Generalised dry skin, keratosis pilaris,sparse eyebrows, curly wooly hair |
| II.3 | 5 years / M | Antenatal data not available | Moderate ID | 95 cms (-3 to -4SD) | Atrial septal defect | Coarse, broad face, downslanting palpebral fissures, bulbous tip of nose. | - | Curly wooly hair |
| II.4 | 8 years / M | Antenatal data not available | Moderate ID | 115 cms (-2 to -3SD) | Hypertrophic cardiomyopathy | Coarse, broad face. downslanting palpebral fissures, bulbous tip of nose. ptosis | - | Curly hair |
| II.5 | 18 months / M | Polyhydramnios | Mild development delay | 73 cms (-3 SD) | Atrial septal defect | Coarse, broad face. downslanting palpebral fissures, bulbous tip of nose. | - | Curly, scant hair |
| III | 15 months / M | Antenatal data not available | Mild development delay | 71 cms (-3 SD) | - | Downslanting palpebral fissures, bulbous tip of nose | - | Multiple café au lait spots, plexiform neurofibroma Heterozygous mutation (c.2033dupC) in exon 18 of NF1 gene |
| IV | 6 year / F | Antenatal data not available | Delayed psychomotor development | 107 cms (5th to 25th centile) | Hypertrophic cardiomyopathy | Coarse facies, broad forehead, depressed nasal bridge, epicanthic folds, hypertelorism, thick, fleshy earlobes, full nasal tip, thick lips |
| Sparse, fine scalp hair, skin - small papilloma at the root of the nasal tip was noticed, hyperkeratosis of palms & soles |
Heart disease was present in 17 out of 20 patients (85%) similar to the estimated frequency between 50 - 80% (Allanson & Roberts, 2011). NS and related disorders are one of the most common syndromic cause of heart defects. Several cardiovascular phenotypes are found, with pulmonary valvular stenosis being the most common in our cohort followed by hypertrophic cardiomyopathy and atrial septal defects.
Neonates with NS usually have normal birth weight and body length. Infants, however have feeding difficulties that result in failure to thrive, most evident in the first year of life. Severe feeding difficulty and gastroesophageal reflux disease were present in three of our patients (Table 2- I.4, I.5, II.1), requiring prolonged gastrostomy in one of them.
In childhood, short stature is almost a universal finding and height usually follows the third centile with an attenuated pubertal spurt. A study reported that 30% of individuals with Noonan syndrome have a height in the normal adult range while 40 – 50% individuals have an adult height below the third centile (Noonan et al., 2003). This may be due to growth hormone deficiency because of neurosecretory dysfunction or growth hormone resistance. The US Food and Drug Administration in 2007 approved Growth Hormone (GH) replacement therapy with recombinant human growth hormone for Noonan Syndrome. Several long and short term studies on the use of GH in different parts of the world reveal significant improvement in the height velocity in children with NS (Tamburrino et al., 2015; Romano et al., 2009; Noordam et al., 2008; Osio et al., 2005; Ogawa et al., 2004).
In our study cohort, sparing an adult and a fetus, short stature was recorded in all the patients. Two patients have been receiving growth hormone therapy for a few years. One of the boys (Table 2- I.9) had height at -5 to - 4 SD at 4.5 years of age, delayed bone age, low IGF-1 levels and inadequate response to clonidine in growth hormone stimulation test. On receiving an average 0.15 units/ kg /day of GH subcutaneously, he showed significant increase in height velocity initially and gained 7.5 cm in the first year of therapy. At 14 years of age, his height is at -2SD from the mean (comparable to the mid parental height centile). For the second boy (Table 2- I.8) who received growth hormone treatment, GH was initiated at 0.15 units/kg/day from 4 years of age. The height increased from -3 SD to -2 SD within 3 years of initiation of therapy. However, it has not increased beyond -2 SD from the mean demonstrating a short-term increase in growth. There were no complications of hypertrophic cardiomyopathy or hematologic disturbances in these patients.
Occasionally NS is referred to as ‘Pseudo-Turner or Male Turner syndrome’, due to similar findings of short stature, webbed neck and lymphedema. Interestingly, we had one girl (Table 2- I.10) who was evaluated for short stature. The karyotype revealed 45,X [3] / 46, XX [47] confirming the diagnosis of mosaic Turner syndrome. However, she also had some characteristic NS-like craniofacial dysmorphism and systemic malformations - epicanthic folds, hypertelorism, low nasal bridge, downward eye slant, low set ears, pulmonary stenosis and lentigines. Molecular panel testing for Noonan-related genes identified a heterozygous mutation (c.2536G>A) in the SOS1 gene that has been implicated in Noonan syndrome.
In most affected individuals of NS, intelligence is within the normal range, with the intelligence quotient ranging from 70-120. Mild to severe learning disability is reported in 25% and 10% of the patients respectively. Furthermore, in literature it has been noted that the verbal performance is significantly lower than the non-verbal performance. In contrast, neurologic abnormalities have been reported to be universally present in CFC and range from mild to severe (Yoon et al., 2007). In our cohort of NS patients, development / intellect was appropriate for age in 54% (7/13) of patients. The children with a clinical diagnosis of CFC syndrome and Costello syndrome in the cohort had global development delay that was of mild to moderate severity.
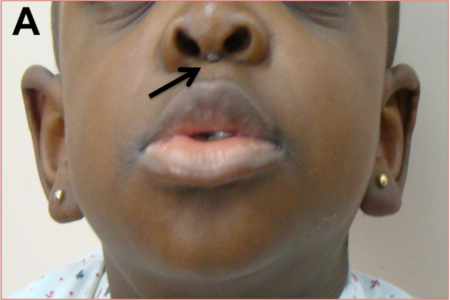
Among the RASopathies, the dermatologic findings are the most common in cardiofaciocutaneous syndrome (CFC). These include xerosis, hyperkeratosis, ichthyosis, keratosis pilaris, ulerythema ophryogenes, eczema, pigmented moles, hemangiomas, and palmoplantar hyperkeratosis. The hair is typically sparse, curly, fine or thick, wooly or brittle; eyelashes and eyebrows may be absent or sparse. Nails may be dystrophic or fast growing. Apart from these similar features, Costello syndrome is characterised by papillomas of face and perianal region, as also found in our patient IV (Fig 3).
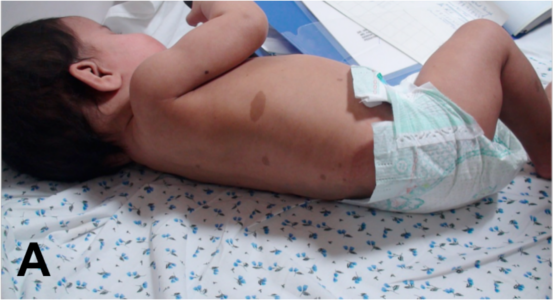
Patients with NF1 have café au lait spots, axillary freckling and neurofibromas in skin. NS may also have skin manifestations, particularly follicular keratosis over extensor surfaces, lentigines and café-au-lait spots. One child in the current cohort (Table 2-III) presented with clinical features characteristic of both neurofibromatosis type 1 and Noonan syndrome (NFNS syndrome- OMIM 601321). The NF1 features at presentation were > 6 café-au-lait spots. Follow up MRI identified cervical plexiform neurofibroma (Fig 4A, 4B). In addition the child had short stature, hypertelorism, ptosis, nystagmus, low-set ears, webbed neck and pectus deformity suggestive of NS. Mutation analysis revealed a heterozygous truncation mutation in NF1:c.2033dupC, thereby confirming the diagnosis of NFNS. The parents were normal on clinical examination. Recently, a similar report of a family with multiple café au lait spots and NS-like facial features in a child (fulfilling criteria of NF1) and mother (not fulfilling criteria for NF1) revealed mutation in MAP2K2 gene (Takenouchi et al., 2014). This gene has been originally implicated in CFC syndrome which further illustrates the phenotypic and genetic overlap in RASopathies.
Individuals with Noonan syndrome have upto three fold increased risk of malignancies which include juvenile myelomonocytic leukemia, acute lymphoblastic leukemia, acute myeloid leukemia and solid tumours such as rhabdomyosarcoma and neuroblastoma (Jongmans et al., 2011; Strullu et al., 2014).
In particular, individuals with germline mutations in the PTPN11 gene have a predisposition to Juvenile myelomonocytic leukemia (JMML) (Strullu et al., 2014). In our cohort, patient I.4 was diagnosed with JMML at 3 months of age. He also had atrioventricular canal defect, feeding difficulties and severe failure to thrive. In view of these, he was clinically suspected and found to be carrying a heterozygous mutation in the PTPN11 gene (c.218C>T, p.T73I), confirming the diagnosis of Noonan syndrome. This mutation has been previously identified in multiple Noonan patients with JMML, with a milder clinical course (Aoki et al., 2008).
For prenatal diagnosis, the ultrasonographic markers for NS are non specific. In the absence of family history NS is not routinely suspected and prenatal testing is not typically offered. In our cohort, the antenatal data of five patients was available and showed abnormalities. One patient (Table 2-I.2) had increased nuchal fold thickness (NFT) of 5.6 mm with femur length at 5th centile and polyhydramnios in the 2nd trimester scan. Fetal chromosomes were tested and were normal. The neonate came to medical attention at 3 months of age for dysmorphism and joint movement restriction.
One fetus (Table 2-I.1) which was terminated in view of increased NFT (5.68 mm), bilateral hypoplastic kidneys and severe pulmonary stenosis on antenatal ultrasound at 18 weeks gestation was clinically diagnosed with Noonan syndrome in view of the facial phenotype.
The other three patients (Table 2-II.1, II.2, II.5) who had antenatal polyhydramnios and unilateral hydronephrosis came to medical attention after birth for developmental delay, facial dysmorphism and congenital heart disease.
The prenatal features described for NS are increased nuchal translucency (NT), distended jugular lymphatic sacs (JLS), cystic hygroma, hydrops fetalis, pleural effusion, polyhydramnios, congenital heart disease and renal abnormalities (Myers et al., 2014).
Out of these, increased NT has the strongest association with Noonan syndrome. However, there is considerable debate as to when to offer prenatal molecular testing for Noonan syndrome, either following a first trimester increased NT or if there are associated anomalies in the 2nd trimester scan with normal fetal chromosomes.
In recent studies in fetuses with an increased NT and a normal karyotype, mutations have been reported in 9–18% of cases. Lee et al. (2009) identified PTPN11 mutations in 2% of fetuses with increased NT and 16% of fetuses with increased NT and cystic hygroma. In another study, in fetuses with increased NT and normal karyotype, PTPN11 and KRAS mutations were found in 15.8%. This group strongly advocated genetic counseling and testing for Noonan syndrome in case of increased NT and normal karyotype, even in the absence of additional associated abnormalities (Houweling et al., 2010). On the other hand, Croonen et al. (2013), based on their mutation detection rate of 17.3% in fetuses with ultrasound findings of increased NT, distended jugular lymphatic sacs (JLS), hydrothorax, renal anomalies, polyhydramnios, cystic hygroma, cardiac anomalies, hydrops fetalis and ascites, recommended prenatal testing of PTPN11, KRAS and RAF1 in pregnancies with an increased NT and at least one of the additional ultrasonologic features.
In conclusion, Noonan syndrome and the other RASopathies have multisystem morbidities. The clinical features are overlapping and there is extensive genetic heterogeneity. In case of antenatal or postnatal clinical suspicion, with availability of next generation panel testing, the genes in the Ras/MAPK pathway implicated with the phenotypes of RASopathies can be tested for confirmation of diagnosis. Furthermore, in view of multisystemic involvement, multidisciplinary management and follow up of diagnosed patients is essential.
1. Allanson JE, Roberts AE. Noonan Syndrome. 2001 Nov 15 [Updated 2011 Aug 4]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1124
2. Aoki Y, et al. The RAS/MAPK Syndromes: Novel Roles of the RAS Pathway in Human Genetic Disorders. Hum Mutat 2008; 29: 992-1006.
3. Aoki Y, et al. Recent advances in RASopathies. J Hum Genet 2016; 61: 33-9. doi: 10.1038/jhg.2015.114.
4. Bezniakow N, et al. The RASopathies as an example of pathway disturbances- Clinical Presentation and Molecular Pathogenesis of selected syndromes. Dev Period Med 2014; 18: 285-296.
5. Croonen EA, et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. Eur J Hum Genet 2013; 21: 936-942.
6. Houweling AC, et al. Prenatal detection of Noonan syndrome by mutation analysis of the PTPN11 and the KRAS genes. Prenat Diagn 2010; 30: 284–286.
7. Jongmans MC, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet 2011; 19: 870-874.
8. Korf B, et al. The Third International Meeting on Genetic Disorders in the RAS/MAPK Pathway: Toward a Therapeutic Approach. Am J Med Genet A 2015; 167: 1741–1746.
9. Lee KA, et al. PTPN11 analysis for the prenatal diagnosis of Noonan syndrome in fetuses with abnormal ultrasound findings. Clin Genet 2009; 75:190–194.
10. Myers A, et al. Perinatal features of the RASopathies: Noonan syndrome, Cardiofaciocutaneous syndrome and Costello syndrome. Am J Med Genet Part A 2014; 164A: 2814–2821.
11. Noordam C, et al. Long-term GH treatment improves adult height in children with Noonan syndrome with and without mutations in protein tyrosine phosphatase, non-receptor-type 11. Eur J Endocrinol 2008; 159: 203–208.
12. Ogawa M, et al. Clinical evaluation of recombinant human growth hormone in Noonan syndrome. Endocr J 2004; 51: 61–68.
13. Osio D, et al. Improved final height with long-term growth hormone treatment in Noonan syndrome. Acta Paediatr 2005; 94: 1232–1237.
14. Romano AA, et al. Growth response, near-adult height, and patterns of growth and puberty in patients with Noonan syndrome treated with growth hormone. J Clin Endocrinol Metab 2009; 94: 2338–2344.
15. Strullu M, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 2014; 51: 689-697.
16. Takenouchi T, et al. Multiple café au lait spots in familial patients with MAP2K2 mutation. Am J Med Genet A.2014; 164A: 392-396.
17. Tamburrino F, et al. Response to long-term growth hormone therapy in patients affected by RAsopathies and growth hormone deficiency: Patterns of growth, puberty and final height data. Am J Med Genet A 2015:167: 2786-294.
18. Tidyman WE, Rauen KA. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009; 19: 230–236.
19. Noonan JA, et al. Adult height in Noonan syndrome. Am J Med Genet A. 2003; 123A: 68–71.
20. Yoon G, et al. Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol 2007; 49: 894–899.
| Abstract | Download PDF |
 A
A 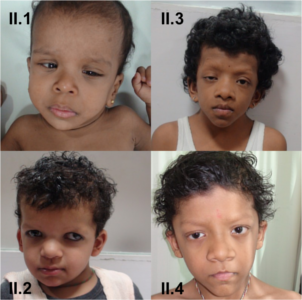 B
B  C
C
 A
A 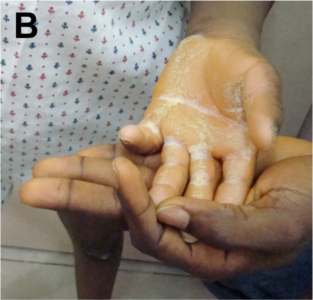 B
B 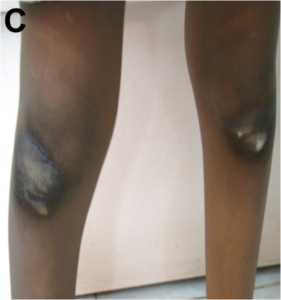 C
C
 A
A 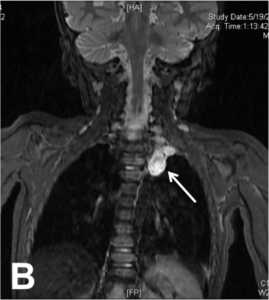 B
B