| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| Type of well defined
leukoencephalopathy | Underlying pathophysiology | Examples
|
| Hypomyelinating disorders | Primary disturbance in the formation of myelin |
|
|
| Secondary to neuron or astrocyte dysfunction (including abnormal interaction between oligodendrocytes and neurons) |
|
| Dysmyelinating disorders (Delayed or disturbed myelination) |
|
|
| Demyelinating disorders (leukodystrophies) |
|
|
| Disorders related to myelin splitting (cystic degeneration of myelin) | With myelin loss |
|
|
| Without myelin loss |
|
| Disorders secondary to axonal damage |
|
|
| Others |
|
|
As per the recent GLIA (Global Leukodystrophy Initiative) Consortium consensus statement, more than 30 distinct leukodystrophy conditions have been characterized, which are listed below in the alphabetical order in Table 2 (Vanderver A et al., 2015).
| Disorder | Gene(s) | Pattern of
inheritance |
| Adrenoleukodystrophy X linked (X-ALD) |
| XL |
| Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia (ALSP):
|
| AD |
| Aicardi– Gouti�res Syndrome (AGS) |
| Usually AR but maybe AD |
| Alexander disease (AxD) |
| AD |
| Autosomal Dominant Leukodystrophy with Autonomic disease (ADLD) |
| AD |
| Canavan disease |
| AR |
| Cerebrotendinous Xanthomatosis (CTX) |
| AR |
| Chloride Ion Channel 2 (ClC-2) related leukoencephalopathy with intramyelinic oedema (leukoencephalopathy with ataxia) |
| AR |
| eIF2B-related disorders (Vanishing White Matter Disease (VWMD) or Childhood ataxia with central nervous system hypomyelination (CACH)) |
| AR |
| Fucosidosis |
| AR |
| Globoid cell Leukodystrophy (Krabbe) |
| AR |
| Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) |
| AD |
| Hypomyelination with brainstem and spinal cord involvement and leg spasticity (HBSL) |
| AR |
| Hypomyelination with congenital cataract (HCC) |
| AR |
| Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) |
| AR |
| Leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) |
| AR |
| Megalencephalic Leukoencephalopathy with subcortical cysts (MLC) |
| AR |
| Metachromatic leukodystrophy (MLD) and its biochemical variants |
| AR |
| Oculodentodigital dysplasia (ODDD) |
| Usually AD maybe AR |
| Pelizaeus Merzbacher disease (PMD) |
| XL |
| Pelizaeus Merzbacher like-disease (PMLD) |
| AR |
| Peroxisomal Biogenesis disorders (including Zelleweger, neonatal Adrenoleukodystrophy and Infantile Refsum) |
| AR |
| Pol-III related disorders (4H syndrome - hypomyelination, hypodontia and hypogonadotropic hypogonadism) |
| AR |
| Polyglucosan Body Disease (PGBD) |
| AR |
| RNAse T2 deficient leukoencephalopathy |
| AR |
| Sialic acid storage disorders (Salla disease, Infantile sialic acid storage disease and Intermediate form) |
| AR |
| Single enzyme deficiencies of peroxisomal fatty acid beta oxidation:
|
| AR |
| Sj�gren–Larsson syndrome |
| AR |
| SOX10-associated PCWH - peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome and Hirschsprung disease |
| AD |
| 18q minus syndrome | ∙ Contiguous gene deletion involving the MBP gene | Majority are de novo; deletion can be inherited |
Leukoencephalopathies include inherited vasculopathies (eg. Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) and COLA1 & COLA2-related disorders), inborn errors of metabolism (e.g. organic acidemias and disorders of aminoacid metabolism), disorders affecting the neurons of the cerebral cortex or other gray matter structures (e.g. infantile variants of GM1 and GM2 gangliosidosis and neuronal ceroid lipofuscinosis), those with both white and gray matter involvement (e.g. mitochondriopathies such as Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) and Myoclonic Epilepsy with Ragged-Red Fibers (MERRF) syndromes, POLG-related disorders and familial hemophagocytic lymphohistiocytosis) and acquired myelin disorders (eg. multiple sclerosis) which may be of infectious or post infectious etiology or due to toxic, hypoxic or non-genetic vascular insults.
There is limited data on the overall prevalence of leukodystrophies and the relative frequencies of different leukodystrophies. Metachromatic leukodystrophy is reported to occur in 1 in 40,000 to 170,000 individuals world-wide (von Figura et al., 2001). Ethnic preponderance has been reported in some leukodystrophies. Canavan disease is relatively common with a high carrier frequency in the Ashkenazi Jewish population. Two common mutations (E285A and Y231X) accounting for 98% of the disease-causing alleles of the ASPA gene (Sistermans et al., 2001). Megalencephalic leukodystrophy with subcortical cysts (MLC) is a common leukodystrophy described in the Agarwal community in India and in a study by Gorospe et al (2004) all 31 cases tested were found to result from a common mutation (320insC) in the MLC1 gene, suggesting a founder effect in this population (Gorospe et al. 2004).
The clinical diagnosis of leukodystrophies and genetic leukoencephalopathies is often challenging due to considerable overlap in the clinical features. Though the advances in recognition of the neuroimaging patterns of these disorders has improved the diagnostic yield, more than half of these disorders still remain undiagnosed.
The clinical features are predominantly neurologic and almost invariably affect the motor system and are progressive in nature. Extra neurologic features provide vital clues to arrive at a specific diagnosis.
The onset of the symptoms is variable ranging from connatal (at birth) to adulthood (Table 3) and most of these disorders present with variable severity across all age groups.
| Disorder | Infantile (first
year) | Late infantile
(1-5yrs) | Juvenile
(5-12yrs) | Adolescent
and adulthood
|
| Metachromatic leukodystrophy (MLD) | √ (most common type of MLD) | √ | √ | |
| Pelizaeus Merzbacher Disease | √ | √ (classic form) | ||
| Krabbe disease | √ (classic form) | √ | √ | √ |
| Alexander disease | √ (most common variant) | √ | √ | √ |
| Canavan disease | √ | |||
| X-linked adrenoleukodystrophy | √ | √ | ||
| Childhood ataxia with central nervous system hypomyelination (CACH) | √ | √ | √ | √ |
| Megalencephalic Leukoencephalopathy with subcortical cysts (MLC) | √ | √ | √ | √ |
| Aicardi–Gouti�res Syndrome (AGS) | √ | |||
| Giant axonal neuropathy type I | √ | |||
| Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) | √ | √ | ||
| Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) | √ | √ | √ | |
| Gradual progressive decline |
|
| Rapid decline |
|
| Episodic decline (triggers may be an event of minor head trauma or febrile illness) |
|
| Macrocephaly
|
|
| Microcephaly
|
|
Associated extraneurologic features provide additional clues to the diagnosis. They are summarised in the following table (Table 6).
| Clinical feature | Disorder | |
| Facies
| ||
| Dysmorphism | 18q microdeletion |
|
| Coarse facies | Sialic acid storage disease, Fucosidosis, Multiple sulfatase deficiency, Mucopolysaccharidosis |
|
| Progeroid appearance | Cockayne syndrome |
|
| Dental anomalies | ||
| Dental anomalies | Oculodentodigital dysplasia (enamel hypoplasia) |
|
| POL III related disorders (not universal and highly variable-oligodontia, hypodontia, delayed eruption, altered sequence of eruption, abnormal colour /shape) |
||
| Cockayne syndrome (propensity for cavities (most common), abnormal shape, hypodontia, oligodontia and enamel hypoplasia) |
||
| Peroxisomal disorders (enamel defects of secondary teeth) |
||
| Eyes
| ||
| Cataracts | At birth | Hypomyelination with congenital cataract (HCC), Childhood ataxia with central nervous system hypomyelination (CACH) (only connatal cases), peroxisomal disorders |
|
| Childhood onset | Cerebrotendinous Xanthomatosis |
| Cherry red spot | Sialidosis |
|
| Glaucoma | Aicardi–Gouti�res Syndrome, Oculodentodigital dysplasia |
|
| Optic atrophy | Metachromatic leukodystrophy |
|
| Retinitis pigmentosa (night blindness) | Refsum disease (adolescent and adults) |
|
| Vascular retinal defects | Cerebroretinal microangiopathy with calcifications and cysts (Coats plus syndrome) |
|
| Glistening white dots in the retina
(perifoveal) | Sjogren Larsson syndrome (pathognomonic in a patient with ichthyosis) |
|
| Nystagmus | Early onset/ congenital | PMD & PMLD (prominent feature) SOX10 related disease |
|
| Later age | Oculodentodigital dysplasia, Pol III related 4H syndrome - hypomyelination, hypodontia and hypogonadotropic hypogonadism),18q del, Alexander, Canavan |
| Skin manifestations
| ||
| Angiokeratoma corporis diffusum | Fucosidosis |
|
| Icthyosis | Congenital | Sjogren Larsson syndrome |
|
| Childhood | Multiple sulfatase deficiency |
|
| Adulthood | Refsum disease |
| Hyperpigmentation | X-ALD/AMN [Figure 1] |
|
| Xanthomas | Cerebrotendinous xanthomatosis |
|
| Photosensitivity | Cockayne, Tay syndrome |
|
| Chilblains | Aicardi– Gouti�res Syndrome |
|
| Endocrinologic manifestations
| ||
| Adrenal insufficiency | X-ALD, peroxisome biogenesis disorders |
|
| Hypothyroidism | 4H syndrome - hypomyelination, hypodontia and hypogonadotropic hypogonadism), Aicardi– Gouti�res Syndrome |
|
| Hypogonadotropic hypogonadism | 4H syndrome - hypomyelination, hypodontia and hypogonadotropic hypogonadism) |
|
| Ovarian dysgenesis (Premature
ovarian failure) | Ovarioleukodystrophy (CACH), AARS2 mutation-related |
|
| Hepatobiliary manifestations
| ||
| Hepatosplenomegaly | Lysosomal storage disorders’s, (multiple sulfatase deficiency, galactosialidosis, sialic acid disorders) |
|
| Hepatic dysfunction | Peroxisomal disorders (isolated hepatomegaly +/- hepatic
dysfunction) |
|
| Gall bladder dysfunction | MLD (causes gall bladder papillomatosis) |
|
| Skeletal system
| ||
| Chondrodysplasia punctata | Peroxisomal disorders |
|
| Dysostosis multiplex | Multiple sulfatase deficiency, sialidosis |
|
| Short stature | Cockayne syndrome, 4H leukodystrophy |
|
| Hearing deficit
| ||
| Hearing impairment (Commonly
central origin-sensorineural) | Peroxisomal biogenesis disorders (early onset) |
|

Other specific sequences include:
Serial imaging with an interval of atleast 6-12 months is pertinent to distinguish between permanent hypomyelination and delayed myelination especially in children less than 2 years of age. CT scan is useful for detecting calcifications.
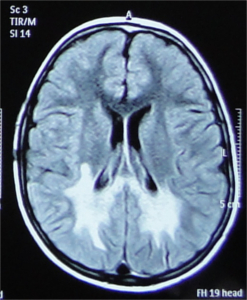
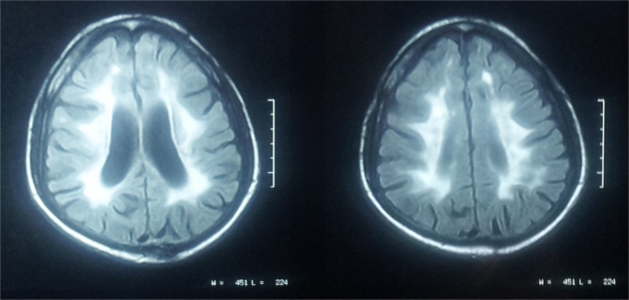
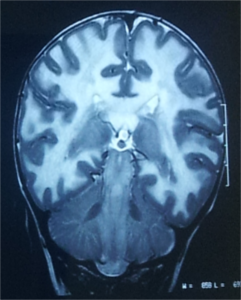
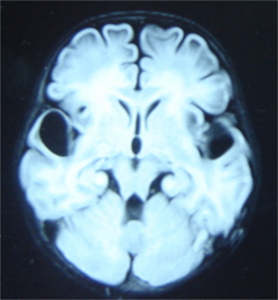
A stepwise approach to recognising the MRI pattern helps in differentiating various white matter disorders
(Schiffmann & van der Knaap, 2009). This involves first recognising whether it is hypomyelination or delayed myelination
or demyelination.
∙ Hypomyelination: Hypomyelination on MRI is characterised by mild T2 hyperintensity in combination with T1
hyperintensity (=normal signal), T1-isointensity or mild T1-hypointensity relative to gray matter structures.
Table 7 lists the conditions with hypomyelination and additional findings which provide a possible clue to
diagnosis.
| With cerebellar involvement (inconstant) | Normal corpus callosum
Thin corpus callosum
|
| With basal ganglia involvement |
|
| With absence of cerebral atrophy (or atrophy in late stages) with normal basal ganglia |
|
| With global atrophy |
|
| Confluent lesions | |
| Predominant | Disorder
|
| localization | |
| Frontal |
|
| Pareito-occipital |
|
| Periventricular |
|
| Subcortical |
|
|
|
| Diffuse cerebral |
|
| Posterior fossa | Cerebellum + middle cerebellar peduncles + Brainstem predominance
|
| Cerebellum and cerebellar peduncles predominance
|
|
| Brainstem predominance
|
|
| Temporal |
|
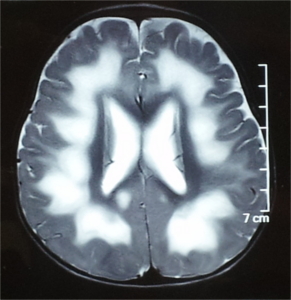
Serial biochemical testing can be done to look for the disease etiology. The type of testing, either biochemical testing or direct single gene testing, may be dictated by the clinical and neuroimaging findings. If a specific etiology cannot be conclusively established by clinical evaluation and imaging features then an approach of ruling out the rapidly diagnosable and those with treatment options may be undertaken. Table 9 lists the biochemical screening tests and the specific target disorders. It is also important to rule out nutritional deficiencies which can cause white matter changes such as vitamin B12 deficiency which can be treated. Some laboratory tests useful in diagnosing genetic white matter disorders are listed below in Table 9.
| Screening test | Disorder
| |
| Enzyme assays | Krabbe (galactosyl cerebrosidase) |
|
| Urinary Analysis | Sulfatides | MLD |
|
| Glysoaminoglycans | Multiple sulfatase deficiency |
|
| Organic acids | L2 glutaric aciduria (↑ concentration of L-2-hydroxyglutaric acid
and lysine) |
|
| Aminoacids | Aminoacidopathies |
| Plasma Very long chain fatty acids | ALD (C26:0, ↑ ratio of C24:0 to C22:0, ↑ ratio of C26:0 to C22:0)
|
|
| Plasma cholestanol | CTX |
|
| Mitochondrial disorders Blood lactate,
pyruvate, aminoacids |
|
|
Additional specific testing can be done at the discretion of the physician and as required, to detect abnormalities which may either provide additional diagnostic clues or may help in the overall patient management such as ophthalmologic evaluation (including slit lamp examination and fundoscopy), hearing evaluation, endocrinologic workup and neurophysiologic studies such as BAER, EMG/NCV,VEP, SSEP) to characterise the involvement of cranial and peripheral nerves (AMN, MLD, Krabbe), optic tracts and spinal tracts.
Molecular diagnostic confirmation can be done by sequence analysis of the relevant gene, based on the recognition of a definitive pattern in MRI or based on the metabolic testing results. However, for many of the leukoencephalopathies, especially the rarer types, there is often a significant overlap in the clinical and neuroimaging features and reliable metabolic testing may not be available; therefore, broad spectrum testing in the form of next generation sequencing-based multigene panel testing for leukodystrophy and genetic leukoencephalopathy genes or whole exome sequencing can be applied to come to a conclusive diagnosis.
Treatment options in general at present are largely symptomatic and supportive, while curative therapies are limited and inadequate. Supportive therapy is aimed at improving the quality of life and involves various strategies, common as well as tailored to individual needs. These include management of spasticity (medications, physiotherapy, orthotics), seizure control and prevention (anticonvulsants), surgical release of contractures and scoliosis correction, gastrostomy for severe dysphagia, proper wheelchair seating, special education, assistive communication devices, and nutritional support.
Genetic counseling is an important component of management of these conditions and the affected families can be offered appropriate counseling about the recurrence risks and about prenatal diagnostic testing options to prevent recurrence.
Specific therapies are available for certain disorders. Early hematopoietic stem cell transplantation (HSCT), though not curative, attenuates the clinical course and prolongs survival of infantile Krabbe disease and X-ALD. Dietary therapy with oral chenodeoxycholic acid (750 mg/day) corrects the biochemical abnormalities and reverses symptoms in Cerebrotendinous xanthomatosis. Hormone therapy can be life saving by preventing Addisonian crisis in susceptible individuals with X-ALD. Early institution (prior to occurrence of MRI abnormalities) of oral Lorenzo’s oil (erucic acid and oleic acid combination) lowers plasma levels of very long chain fatty acids (VLCFA) in patients with X-ALD. Early recognition can be beneficial in those conditions with definite treatment options, and on the whole, preventive, symptomatic and supportive care with multidisciplinary involvement, are of prime importance in the management of patients with genetic white matter disorders.
1. Di Rocco M, et al. Genetic disorders affecting white matter in the pediatric age. Am J Med Genet B Neuropsychiatr Genet. 2004; 129B: 85-93.
2. Gorospe JR, et al. Indian Agarwal megalencephalic leukodystrophy with cysts is caused by a common MLC1 mutation. Neurology 2004; 62: 878-82.
3. Parikh S, et al, A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephalopathies. Mol Genet Metab 2015; 114: 501-515.
4. Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72:750-759.
5. Sistermans EA, et al. Mutation detection in the aspartoacylase gene in 17 patients with Canavan disease: four new mutations in the non-Jewish population. Europ J Hum Genet 2000; 8: 557-560.
6. van der Knaap MS. Magnetic resonance in childhood white matter disorders. Dev Med Child Neurol 2001; 43:705-712.
7. Vanderver A, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015; 114: 494-550.
8. von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001: 3695-3724.
| Abstract | Download PDF |